

RCBTB1基因相關遺傳性視網膜病變是一種近年發現的極為罕見的遺傳性視網膜疾病(IRD)。RCBTB1致病基因的突變可導致早發型視網膜色素變性、遲發型脈絡膜視網膜萎縮等多種IRD臨床表型。RCBTB1基因相關視網膜病變的遺傳方式為常染色體隱性遺傳。RCBTB1基因在維持視網膜色素上皮細胞線粒體功能及抗氧化應激防御機制中發揮重要作用。未來需進一步確定在RCBTB1基因相關遺傳性視網膜病變的發病年齡或多器官受累是否存在基因型與表型的相關性,評估腺相關病毒介導的RCBTB1基因替代療法在動物模型中的安全性和有效性,以期發現基因替代治療及干細胞療法的可行性。
引用本文: 黃智琴, 金子兵. RCBTB1基因相關遺傳性視網膜病變的分子診斷與治療策略的研究進展. 中華眼底病雜志, 2024, 40(6): 472-477. doi: 10.3760/cma.j.cn511434-20230921-00394 復制
版權信息: ?四川大學華西醫院華西期刊社《中華眼底病雜志》版權所有,未經授權不得轉載、改編
RCBTB1基因相關遺傳性視網膜病變是一種近年發現的極為罕見的遺傳性視網膜疾病(IRD),至今,Wu等[1]研究報道攜帶雜合子截短突變的2個常染色體顯性家系,全球共報道了來自11個家系的15例攜帶RCBTB1基因純合子或復合雜合突變的IRD患者(圖1,表1)。該疾病由位于染色體13q14.2上的RCBTB1基因突變所致,臨床異質性極其顯著。部分患者可僅表現為早發型視網膜色素變性(RP)、遲發型進展性脈絡膜視網膜萎縮等異常眼底特征[2-4],部分患者除眼部體征外,亦可并發甲狀腺腫大、智力障礙、繼發性閉經、原發性卵巢功能不全等疾病[5]。鑒于該基因突變所致眼病表現各異,對患者進行準確的臨床診斷顯得尤為困難,眼科醫生較易將該類患者誤診甚至漏診。由于在眼科領域針對RCBTB1基因的研究較少,其分子水平的致病機制尚不明確,針對該類罕見IRD的治療手段探究面臨巨大挑戰。現就RCBTB1基因相關遺傳性視網膜病變的分子診斷與治療策略的研究進展作一綜述。
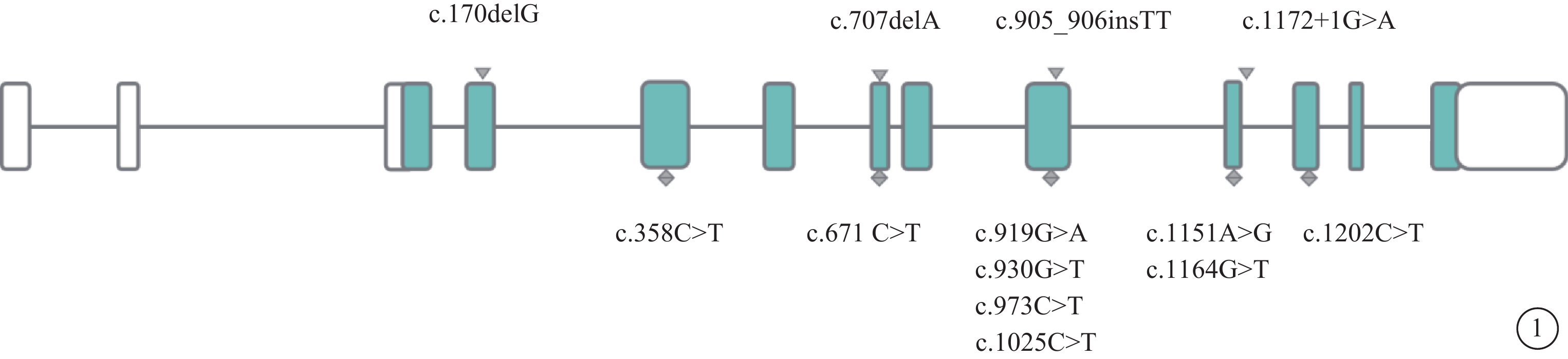
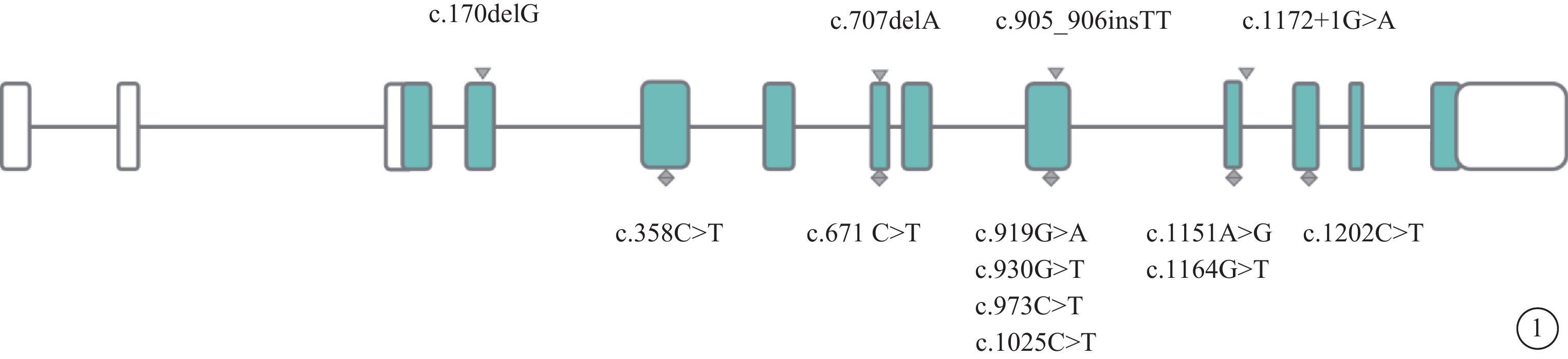 圖1
已報道的RCBTB1基因變異位點
圖1
已報道的RCBTB1基因變異位點
1 臨床特征
目前全球已發表的文獻呈現了RCBTB1基因相關遺傳性視網膜病變的不同臨床表型特征;其中一種表現為家族性滲出性玻璃體視網膜病變(FEVR)、Coats病等視網膜血管性病變(涉及來自2個家系的2例患者)[1];另外一種表現為脈絡膜視網膜病變或RP等視網膜變性病變(包括來自11個家系的15例患者),其中11例患者表現為由黃斑區向周邊區進展的漸進性遲發型脈絡膜視網膜萎縮(平均發病年齡33~62歲),而其他4例表現為早發型RP(平均發病年齡為16歲)[2-5]。眼底自發熒光檢查及定量分析發現,視網膜色素上皮(RPE)萎縮灶擴展的速度(1.0~1.3 mm2/年)[3]與在年齡相關性黃斑變性患者中“地圖樣”RPE萎縮灶進展速度(1.0~2.6 mm2/年)較為接近[6]。隨后Catomeris等[4]亦在RCBTB1基因突變患者中觀察到了相似的RPE萎縮灶進展速率(1.0~1.3 mm2/年)。Coppieters等[5]報道的6個家系中,其中5個家系與Huang等[3]及Catomeris等[4]報道的患者有相同的眼底表型,另1個家系則表現為雙眼RP,與Yang等[2]報道的病例特征一致。值得注意的是,以上家系中的無一病例表現為Wu等[1]報道的Coats病、FEVR(表1)。此外,其他團隊均未報道Coppieters等[5]RCBTB1基因純合錯義變異患者甲狀腺、卵巢、耳部、關節、大腦、肺部等器官的異常改變。光相干斷層掃描和眼底自發熒光檢查可見萎縮性視網膜病變中RPE層及相鄰橢圓體帶的漸進性缺損。由于目前眼底成像數據均顯示RPE萎縮病灶不斷擴大和廣泛的RPE不規則性,推測原發病灶可能位于RPE內,隨后造成嚴重的RPE及光感受器細胞損傷。以往對RCBTB1基因相關性視網膜病變的臨床特征描述亦強調了進一步研究RCBTB1基因在視網膜細胞中的作用的必要性。
2 遺傳模式歸納
2016年,Wu等[1]首次報道中國臺灣地區2個RCBTB1基因雜合移碼突變引起的常染色體顯性遺傳FEVR和Coats病家系。比利時研究團隊報道,來自不同種族的6個家系中RCBTB1基因純合子錯義變異可導致多種臨床表型[5]。近年以來,筆者團隊發表關于RCBTB1基因復合雜合突變c.170delG(p. Gly57Glufs*12)和c.707delA(p. Asn236Thrfs*11)引起脈絡膜視網膜萎縮的病例報道[3]。隨后Catomeris等[4]研究報道,加拿大3個散發病例中RCBTB1基因上的新的純合子錯義變異可引起相似脈絡膜視網膜萎縮性眼底改變[4]。我國張清炯教授團隊通過全基因組測序及靶向外顯子測序,在一散發RP家系中檢測到RCBTB1基因上的復合雜合突變,然而在健康家系中亦能檢測到雜合子截短突變,推斷RCBTB1雙等位基因上均發生突變方可導致疾病的發生和發展,提示RCBTB1相關視網膜病變的遺傳方式為常染色體隱性遺傳[7],這與Wu等[1]的研究發現不一致。3型線粒體視網膜病變與RCBTB1基因相關視網膜病變眼底表型有高度相似性,這兩種視網膜病變均表現為開始于視盤周圍區域的脈絡膜視網膜萎縮,這提示線粒體突變或缺失與RCBTB1基因功能障礙之間可能存在共用的病理生理途徑[8]。
3 RPE細胞模型的建立
干細胞技術被認為是近二十年科學研究領域的重大突破,通過強制表達特定轉錄因子,誘導多能干細胞(iPSC)可以從多種類型的人體體細胞中重新編程,包括成纖維細胞、骨髓間充質基質細胞、黑色素細胞、角質細胞、血液細胞和尿液細胞[9-12]。既往研究中,利用RCBTB1基因突變患者的皮膚成纖維細胞作為iPSC生產的細胞來源,采用了附加體載體重編程試劑盒,這種非整合、非病毒系統,將重編程因子輸送和轉導到患者源的成纖維細胞中,該方法具有相對較高的效率和安全性[13]。到目前為止,基于各種重編程方法從人體體細胞中生成iPSC細胞系已變得更加高效和安全,以便進一步進行臨床應用。以往研究證明,iPSC在長期培養(>50次傳代)后仍能保持多能性,不出現核型異常或多能性喪失。筆者團隊曾使用最多達38次傳代的iPSC進行分化成RPE細胞的實驗,其效率與低傳代(20~30次傳代)的iPSC相似[13]。考慮到可能保留了成纖維細胞的表觀遺傳特征,為進一步驗證從成纖維細胞中重編程的iPSC,還可以進行表觀遺傳和表型分析,這可能解釋不同iPSC細胞系之間存在的差異。盡管目前表觀遺傳分析尚未被列為iPSC重編程質量控制的必要步驟,但當涉及到利用多個細胞系評估細胞系間表型差異時仍應予以關注。
為進一步建立RCBTB1基因相關遺傳性視網膜病變疾病模型,既往研究成功將RCBTB1基因突變患者來源的iPSC誘導分化為RPE細胞。迄今為止,攜帶RCBTB1基因變異的患者在臨床上表現出多種RCBTB1基因相關視網膜病變的類型,包括早發型RP、遲發型漸進性脈絡膜萎縮及玻璃體視網膜疾病。1例來自新加坡的攜帶RCBTB1基因突變的患者近5年隨訪期間的臨床數據不僅記錄了發生在該患者眼底脈絡膜視網膜萎縮進展的自然病程,亦通過大量詳盡的臨床數據分析得出該疾病發病病灶很可能位于RPE層內這一猜想[3]。最近,Carron等[14]也在RCBTB1基因敲除蛙模型中發現其RPE層結構較對照組的RPE層結構更為紊亂,進一步表明RPE細胞在RCBTB1基因相關的視網膜病變中首先并主要受到影響。與臨床觀察結果一致,對比遲發型脈絡膜萎縮患者與健康對照組來源的RPE。筆者團隊研究發現,患者iPSC誘導分化得到的RPE細胞顯示出病理性改變,包括具有短纖毛(0.5~1.0 μm)的RPE細胞比例增加,表面微絨毛密度降低。透射電子顯微鏡可見患者iPSC來源RPE較薄的RPE細胞層及較短的微絨毛結構,還證明患者來源iPSC-RPE的線粒體形態和結構的破壞[15-16],這提示線粒體功能障礙可能在RCBTB1基因缺陷RPE細胞的疾病發病機制中發揮重要作用。然而,RCBTB1蛋白與線粒體的關系以及對細胞能量代謝的影響仍有待進一步研究,將有助于理解其在視網膜病變中的作用。
4 疾病機制探究
RCBTB1基因位于染色體13q14.2上,由13個外顯子組成。該基因在人體各組織廣泛表達,在視網膜及腦組織中表達水平最高。該基因所編碼的RCBTB1蛋白由531個氨基酸組成,具有兩個功能域,一個N-末端染色體濃縮調節因子1(RCC1)-類似域和兩個C-末端的BTB域。最近的研究證明原型RCC1蛋白質作為Ras相關核三磷酸鳥苷酶的鳥嘌呤核苷酸交換因子,在調控細胞周期、DNA損傷/修復及細胞凋亡方面起著至關重要的作用[17-18]。根據已知的蛋白質結構特征,將包含RCC1結構域蛋白質分為5組,其中RCBTB亞組包括RCBTB1、RCBTB2和Bruton酪氨酸激酶抑制因子(IBtk)。IBtk特異性地在淋巴細胞中表達,而RCBTB1和RCBTB2則在所有細胞類型中廣泛表達,并被認為在細胞功能中發揮重要作用[18]。最早關于RCBTB1基因的研究結果提示,其可能在白血病發病機制中發揮腫瘤抑制因子的作用[19]。隨后的研究證明,過表達RCBTB1基因可導致嚙齒動物動脈平滑肌細胞和正常人上皮細胞的細胞肥大,這一生物學過程可能涉及磷脂酰肌醇3激酶或活性氧(ROS)依賴的磷酸酶B途徑的激活[20]。Wu等[1]通過眼底彩色照相檢查在攜帶RCBTB1基因突變的患者中發現異常眼底表現,眼科領域才開始針對RCBTB1基因相關遺傳性視網膜病變的分子遺傳特征、發病機制及相關治療策略進行深入的研究。關于RCBTB1基因突變引起視網膜疾病的致病機制,目前主要存在兩種理論:(1)Wu等[1]提出的單倍劑量不足機制,即由于患者僅攜帶一份正常RCBTB1基因因此體內RCBTB1基因表達減少,從而導致RCBTB1蛋白合成異常。RCBTB1蛋白合成不足可能影響諾里病蛋白/β-連環蛋白(Norrin/β-catenin)信號通路的激活,從而導致視網膜血管生成異常。在嗎琳環介導RCBTB1基因敲除斑馬魚疾病模型中發現其眼內血管生成異常,此外在一種主要用于RPE研究的人RPE細胞系ARPE-19細胞中,發現RCBTB1基因與Norrin/β-catenin信號通路的激活受損有關[1];(2)由Plafker等[21]提出的RCBTB1基因突變導致泛素化受損理論,核因子E2相關因子2,又稱核因子紅細胞衍生2樣2 (Nrf2),這一轉錄因子對于細胞的抗氧化應激反應至關重要,它的活性受到泛素化機制的調控。既往報道,個別與泛素化調控機制相關的基因如KLHL7和TOPORS基因與IRD的致病機制有關[22-23]。Plafker等[21]利用酵母菌雙雜合系統發現RCBTB1蛋白很可能是CUL3-泛素E3連接酶復合物的組成部分。研究報道,2例RCBTB1基因突變患者的外周血單核細胞中NFE2L2通路相關的基因表達水平顯著降低[5]。健康對照來源的iPSC-RPE細胞中證實了RCBTB1蛋白與CUL3-泛素E3連接酶復合物存在相互作用[15]。研究報道,在RCBTB1基因突變患者來源的iPSC-RPE細胞中檢測到與NFE2L2通路相關的基因表達水平較對照組顯著降低。
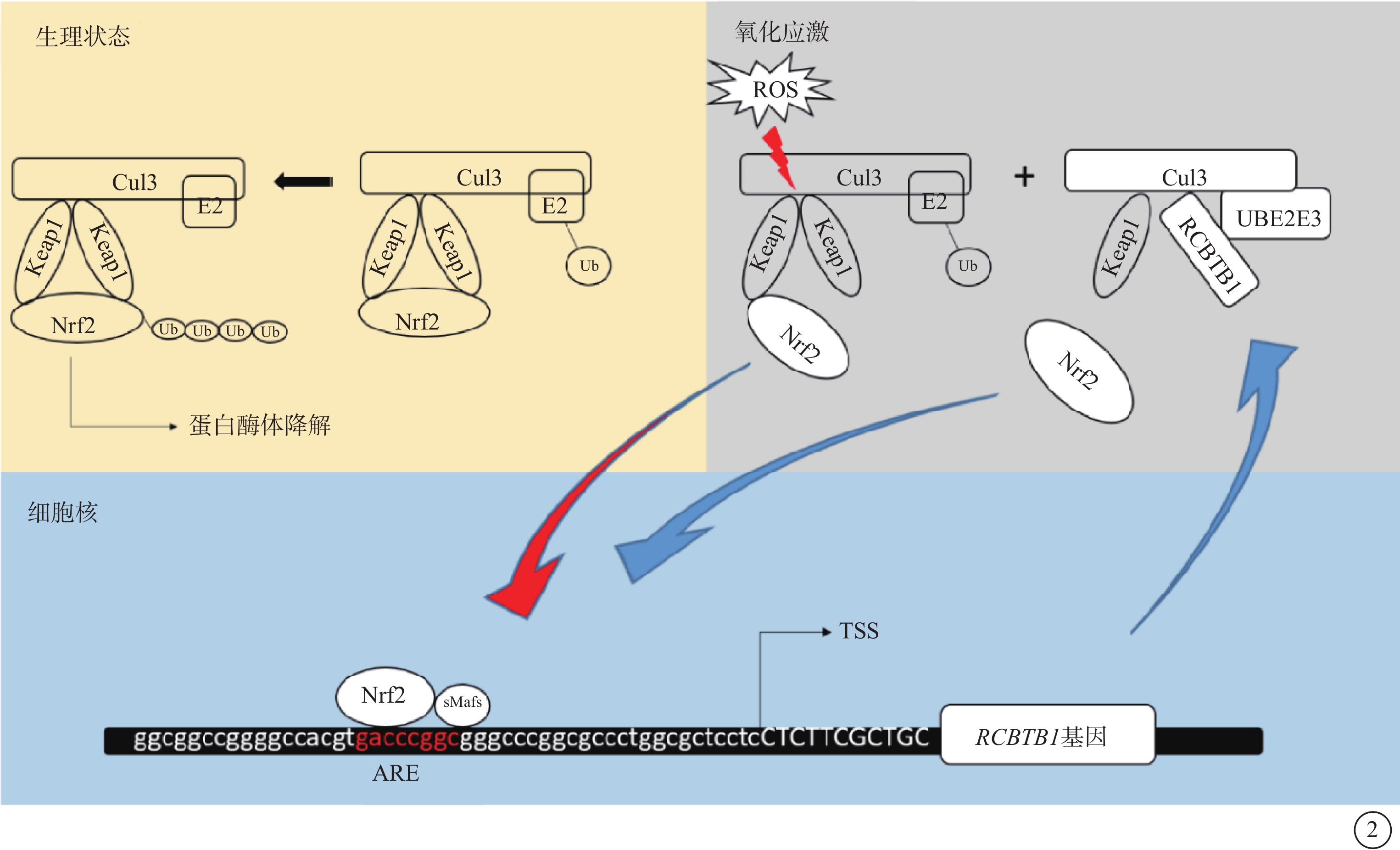
研究表明,RPE細胞可以通過激活NFE2L2通路增強抗氧化防御以減少由活性氧(ROS)引起的細胞損害[24-25]。筆者團隊近來在患者來源iPSC-RPE中發現即使在低濃度過氧叔丁醇(tBHP),一種常用的氧化劑,刺激下也產生大量ROS,表明RCBTB1基因缺陷的RPE細胞比對照組更容易受到tBHP誘導的氧化應激的影響[15],這一發現與先前的研究結果一致[14]。tBHP處理后的對照RPE細胞中RCBTB1蛋白表達水平顯著上調,這表明RCBTB1在RPE細胞氧化應激響應中起著一定作用。鑒于NFE2L2結合的底物適配蛋白Kelch樣環氧氯丙烷相關蛋白1(Keap1)是一種BTB-Kelch蛋白,通過與CUL3-泛素E3連接酶復合物結合來發揮功能,猜測底物適配蛋白RCBTB1和Keap1之間在CUL3-泛素E3連接酶復合物上的競爭解釋RCBTB1在調控NFE2L2水平中的作用。此外,NFE2L2轉錄因子通過結合其下游靶基因啟動子中的抗氧化應答元素(ARE)序列(TGACXXXGC)啟動抗氧化機制,而在RCBTB1啟動子位于轉錄起始位點上游33個核苷酸處存在一個ARE序列。綜上表明,在氧化應激條件下,NFE2L2可能作為正反饋回路的一部分,通過激活RCBTB1基因表達來發揮作用(圖2)。
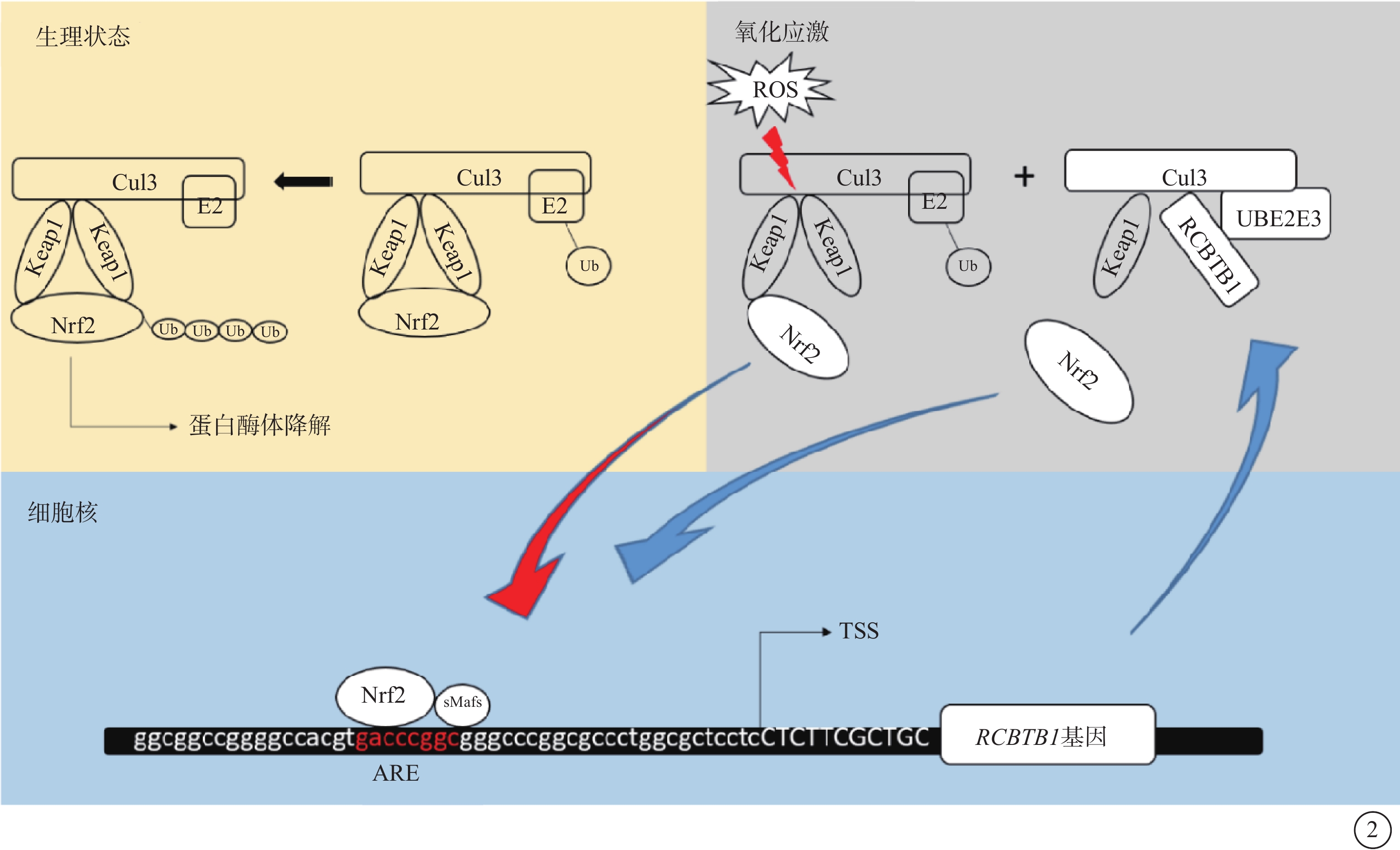 圖2
RCBTB1基因突變可能致病機制示意圖
圖2
RCBTB1基因突變可能致病機制示意圖
Cul3:Cullin3蛋白;E2:泛素連接酶;UBE2E3:泛素結合酶;Keap1:Kelch樣環氧氯丙烷相關蛋白1;Nrf2:核因子紅細胞衍生2樣;Ub:泛素;ROS:活性氧;TSS:轉錄起始位點;ARE:抗氧化應答元素;sMafs:小Maf蛋白
5 治療策略
針對早期發病表現為RP的RCBTB1基因相關遺傳性視網膜病變患者,基因治療應作為首選治療手段。早期病變患者仍保留部分尚未凋亡的RPE細胞及光感受器細胞,通過腺相關病毒(AAV)介導在視網膜下腔或玻璃體中導入攜帶有正常RCBTB1基因片段以替代突變的基因片段,理論上可以修復因RCBTB1蛋白表達不足或者缺失引起的視網膜功能障礙,改善患者視功能。由于在分子遺傳學領域取得的快速進展以及優化的基因傳遞載體的轉導效率,基因治療已經成為個性化治療遺傳性視網膜病變的重要手段。AAV在iPSC-RPE體外轉導過程和RPE的體內轉導表現出相似的轉導效率,這提示iPSC-RPE這一體外細胞模型在評估體內基因傳遞效力時具備實用性[26-27]。含有RCBTB1基因互補DNA(轉錄變體1,NM_018191)的AAV2/2和AAV2/8載體可用于高效轉導iPSC-RPE,并且AAV治療可實現RCBTB1基因互補DNA的長期活化水平表達,支持AAV基因療法在未來臨床試驗中提供持久療效的潛力[16]。然而考慮到RCBTB1高度表達可能會導致RPE細胞中NFE2L2激活通路的敏感性增高,這一點應在未來進行臨床基因治療中進行充分評估。
針對晚期發病表現為漸進性遲發型脈絡膜視網膜萎縮特征的患者,細胞替代治療更為合理,通過移植健康的iPSC-RPE移植片或者iPSC-RPE細胞懸浮液以替代原有的攜帶有RCBTB1基因突變的功能損害的RPE細胞,具備理論可行性。至于RPE細胞的純化、篩選以及如何將RPE移植片或懸浮液有效導入視網膜組織這些問題仍有待解決,需要研究學者與臨床工作者共同努力。多能干細胞移植的不同策略,包括單純的細胞移植、細胞外囊泡和細胞因子釋放,為未來干細胞移植研究方向提供了思路,包括改進細胞移植策略、提高細胞分化效率、解決免疫排斥問題以及推動治療方法的臨床應用[28]。
盡管基因替代RCBTB1基因治療的效果及將iPSC誘導分化為視網膜細胞的穩定與否仍然需要進一步研究,就目前而言,基因療法及干細胞療法仍是治療RCBTB1基因相關遺傳性視網膜病變這一類由明確基因突變引起的遺傳性眼病的主流手段。既往研究表明,N-乙酰半胱氨酸抗氧化劑,可以降低氧化應激刺激條件下RPE細胞中約16%的ROS自由基水平,這提示抗氧化劑在治療RCBTB1基因相關的視網膜病變中可能起到一定療效[15]。
6 小結與展望
本研究歸納總結了來自不同種族的RCBTB1基因相關遺傳性視網膜病變患者的眼部特征及遺傳學特征,提示RCBTB1基因相關視網膜病變的遺傳方式為常染色體隱性遺傳。基于既往研究成果對RCBTB1基因在視網膜細胞中的功能影響及治療策略展開分析,推測其在維持RPE細胞線粒體功能及抗氧化應激防御機制中發揮重要作用,因而應用基因替代治療及干細胞療法治療RCBTB1基因相關遺傳性視網膜病變具備可行性。本研究為探索此類罕見遺傳性視網膜病變的發病機制提供研究思路,為RCBTB1的基因治療及干細胞相關研究提供重要參考,亦為RCBTB1基因治療盡早進入臨床試驗奠定基礎。未來為確定在RCBTB1相關視網膜病變的發病年齡或多器官受累是否存在基因型與表型的相關性,仍需收集更多攜帶RCBTB1基因突變的IRD病例,通過進一步的研究闡明RCBTB1基因在視網膜中的作用,如通過檢測RCBTB1缺陷的視網膜細胞中線粒體DNA拷貝數及抗氧化系統組分(如超氧化物歧化酶2、超氧化物歧化酶1和過氧化氫酶)的表達水平,以研究RCBTB1基因與線粒體的潛在關聯,更好地了解RCBTB1基因在氧化應激應答中的作用。此外,還需要評估AAV介導的RCBTB1基因替代療法在動物模型中的安全性和有效性,以期發現基因替代治療及干細胞療法治療的可行性。
RCBTB1基因相關遺傳性視網膜病變是一種近年發現的極為罕見的遺傳性視網膜疾病(IRD),至今,Wu等[1]研究報道攜帶雜合子截短突變的2個常染色體顯性家系,全球共報道了來自11個家系的15例攜帶RCBTB1基因純合子或復合雜合突變的IRD患者(圖1,表1)。該疾病由位于染色體13q14.2上的RCBTB1基因突變所致,臨床異質性極其顯著。部分患者可僅表現為早發型視網膜色素變性(RP)、遲發型進展性脈絡膜視網膜萎縮等異常眼底特征[2-4],部分患者除眼部體征外,亦可并發甲狀腺腫大、智力障礙、繼發性閉經、原發性卵巢功能不全等疾病[5]。鑒于該基因突變所致眼病表現各異,對患者進行準確的臨床診斷顯得尤為困難,眼科醫生較易將該類患者誤診甚至漏診。由于在眼科領域針對RCBTB1基因的研究較少,其分子水平的致病機制尚不明確,針對該類罕見IRD的治療手段探究面臨巨大挑戰。現就RCBTB1基因相關遺傳性視網膜病變的分子診斷與治療策略的研究進展作一綜述。
圖1
已報道的RCBTB1基因變異位點
1 臨床特征
目前全球已發表的文獻呈現了RCBTB1基因相關遺傳性視網膜病變的不同臨床表型特征;其中一種表現為家族性滲出性玻璃體視網膜病變(FEVR)、Coats病等視網膜血管性病變(涉及來自2個家系的2例患者)[1];另外一種表現為脈絡膜視網膜病變或RP等視網膜變性病變(包括來自11個家系的15例患者),其中11例患者表現為由黃斑區向周邊區進展的漸進性遲發型脈絡膜視網膜萎縮(平均發病年齡33~62歲),而其他4例表現為早發型RP(平均發病年齡為16歲)[2-5]。眼底自發熒光檢查及定量分析發現,視網膜色素上皮(RPE)萎縮灶擴展的速度(1.0~1.3 mm2/年)[3]與在年齡相關性黃斑變性患者中“地圖樣”RPE萎縮灶進展速度(1.0~2.6 mm2/年)較為接近[6]。隨后Catomeris等[4]亦在RCBTB1基因突變患者中觀察到了相似的RPE萎縮灶進展速率(1.0~1.3 mm2/年)。Coppieters等[5]報道的6個家系中,其中5個家系與Huang等[3]及Catomeris等[4]報道的患者有相同的眼底表型,另1個家系則表現為雙眼RP,與Yang等[2]報道的病例特征一致。值得注意的是,以上家系中的無一病例表現為Wu等[1]報道的Coats病、FEVR(表1)。此外,其他團隊均未報道Coppieters等[5]RCBTB1基因純合錯義變異患者甲狀腺、卵巢、耳部、關節、大腦、肺部等器官的異常改變。光相干斷層掃描和眼底自發熒光檢查可見萎縮性視網膜病變中RPE層及相鄰橢圓體帶的漸進性缺損。由于目前眼底成像數據均顯示RPE萎縮病灶不斷擴大和廣泛的RPE不規則性,推測原發病灶可能位于RPE內,隨后造成嚴重的RPE及光感受器細胞損傷。以往對RCBTB1基因相關性視網膜病變的臨床特征描述亦強調了進一步研究RCBTB1基因在視網膜細胞中的作用的必要性。
2 遺傳模式歸納
2016年,Wu等[1]首次報道中國臺灣地區2個RCBTB1基因雜合移碼突變引起的常染色體顯性遺傳FEVR和Coats病家系。比利時研究團隊報道,來自不同種族的6個家系中RCBTB1基因純合子錯義變異可導致多種臨床表型[5]。近年以來,筆者團隊發表關于RCBTB1基因復合雜合突變c.170delG(p. Gly57Glufs*12)和c.707delA(p. Asn236Thrfs*11)引起脈絡膜視網膜萎縮的病例報道[3]。隨后Catomeris等[4]研究報道,加拿大3個散發病例中RCBTB1基因上的新的純合子錯義變異可引起相似脈絡膜視網膜萎縮性眼底改變[4]。我國張清炯教授團隊通過全基因組測序及靶向外顯子測序,在一散發RP家系中檢測到RCBTB1基因上的復合雜合突變,然而在健康家系中亦能檢測到雜合子截短突變,推斷RCBTB1雙等位基因上均發生突變方可導致疾病的發生和發展,提示RCBTB1相關視網膜病變的遺傳方式為常染色體隱性遺傳[7],這與Wu等[1]的研究發現不一致。3型線粒體視網膜病變與RCBTB1基因相關視網膜病變眼底表型有高度相似性,這兩種視網膜病變均表現為開始于視盤周圍區域的脈絡膜視網膜萎縮,這提示線粒體突變或缺失與RCBTB1基因功能障礙之間可能存在共用的病理生理途徑[8]。
3 RPE細胞模型的建立
干細胞技術被認為是近二十年科學研究領域的重大突破,通過強制表達特定轉錄因子,誘導多能干細胞(iPSC)可以從多種類型的人體體細胞中重新編程,包括成纖維細胞、骨髓間充質基質細胞、黑色素細胞、角質細胞、血液細胞和尿液細胞[9-12]。既往研究中,利用RCBTB1基因突變患者的皮膚成纖維細胞作為iPSC生產的細胞來源,采用了附加體載體重編程試劑盒,這種非整合、非病毒系統,將重編程因子輸送和轉導到患者源的成纖維細胞中,該方法具有相對較高的效率和安全性[13]。到目前為止,基于各種重編程方法從人體體細胞中生成iPSC細胞系已變得更加高效和安全,以便進一步進行臨床應用。以往研究證明,iPSC在長期培養(>50次傳代)后仍能保持多能性,不出現核型異常或多能性喪失。筆者團隊曾使用最多達38次傳代的iPSC進行分化成RPE細胞的實驗,其效率與低傳代(20~30次傳代)的iPSC相似[13]。考慮到可能保留了成纖維細胞的表觀遺傳特征,為進一步驗證從成纖維細胞中重編程的iPSC,還可以進行表觀遺傳和表型分析,這可能解釋不同iPSC細胞系之間存在的差異。盡管目前表觀遺傳分析尚未被列為iPSC重編程質量控制的必要步驟,但當涉及到利用多個細胞系評估細胞系間表型差異時仍應予以關注。
為進一步建立RCBTB1基因相關遺傳性視網膜病變疾病模型,既往研究成功將RCBTB1基因突變患者來源的iPSC誘導分化為RPE細胞。迄今為止,攜帶RCBTB1基因變異的患者在臨床上表現出多種RCBTB1基因相關視網膜病變的類型,包括早發型RP、遲發型漸進性脈絡膜萎縮及玻璃體視網膜疾病。1例來自新加坡的攜帶RCBTB1基因突變的患者近5年隨訪期間的臨床數據不僅記錄了發生在該患者眼底脈絡膜視網膜萎縮進展的自然病程,亦通過大量詳盡的臨床數據分析得出該疾病發病病灶很可能位于RPE層內這一猜想[3]。最近,Carron等[14]也在RCBTB1基因敲除蛙模型中發現其RPE層結構較對照組的RPE層結構更為紊亂,進一步表明RPE細胞在RCBTB1基因相關的視網膜病變中首先并主要受到影響。與臨床觀察結果一致,對比遲發型脈絡膜萎縮患者與健康對照組來源的RPE。筆者團隊研究發現,患者iPSC誘導分化得到的RPE細胞顯示出病理性改變,包括具有短纖毛(0.5~1.0 μm)的RPE細胞比例增加,表面微絨毛密度降低。透射電子顯微鏡可見患者iPSC來源RPE較薄的RPE細胞層及較短的微絨毛結構,還證明患者來源iPSC-RPE的線粒體形態和結構的破壞[15-16],這提示線粒體功能障礙可能在RCBTB1基因缺陷RPE細胞的疾病發病機制中發揮重要作用。然而,RCBTB1蛋白與線粒體的關系以及對細胞能量代謝的影響仍有待進一步研究,將有助于理解其在視網膜病變中的作用。
4 疾病機制探究
RCBTB1基因位于染色體13q14.2上,由13個外顯子組成。該基因在人體各組織廣泛表達,在視網膜及腦組織中表達水平最高。該基因所編碼的RCBTB1蛋白由531個氨基酸組成,具有兩個功能域,一個N-末端染色體濃縮調節因子1(RCC1)-類似域和兩個C-末端的BTB域。最近的研究證明原型RCC1蛋白質作為Ras相關核三磷酸鳥苷酶的鳥嘌呤核苷酸交換因子,在調控細胞周期、DNA損傷/修復及細胞凋亡方面起著至關重要的作用[17-18]。根據已知的蛋白質結構特征,將包含RCC1結構域蛋白質分為5組,其中RCBTB亞組包括RCBTB1、RCBTB2和Bruton酪氨酸激酶抑制因子(IBtk)。IBtk特異性地在淋巴細胞中表達,而RCBTB1和RCBTB2則在所有細胞類型中廣泛表達,并被認為在細胞功能中發揮重要作用[18]。最早關于RCBTB1基因的研究結果提示,其可能在白血病發病機制中發揮腫瘤抑制因子的作用[19]。隨后的研究證明,過表達RCBTB1基因可導致嚙齒動物動脈平滑肌細胞和正常人上皮細胞的細胞肥大,這一生物學過程可能涉及磷脂酰肌醇3激酶或活性氧(ROS)依賴的磷酸酶B途徑的激活[20]。Wu等[1]通過眼底彩色照相檢查在攜帶RCBTB1基因突變的患者中發現異常眼底表現,眼科領域才開始針對RCBTB1基因相關遺傳性視網膜病變的分子遺傳特征、發病機制及相關治療策略進行深入的研究。關于RCBTB1基因突變引起視網膜疾病的致病機制,目前主要存在兩種理論:(1)Wu等[1]提出的單倍劑量不足機制,即由于患者僅攜帶一份正常RCBTB1基因因此體內RCBTB1基因表達減少,從而導致RCBTB1蛋白合成異常。RCBTB1蛋白合成不足可能影響諾里病蛋白/β-連環蛋白(Norrin/β-catenin)信號通路的激活,從而導致視網膜血管生成異常。在嗎琳環介導RCBTB1基因敲除斑馬魚疾病模型中發現其眼內血管生成異常,此外在一種主要用于RPE研究的人RPE細胞系ARPE-19細胞中,發現RCBTB1基因與Norrin/β-catenin信號通路的激活受損有關[1];(2)由Plafker等[21]提出的RCBTB1基因突變導致泛素化受損理論,核因子E2相關因子2,又稱核因子紅細胞衍生2樣2 (Nrf2),這一轉錄因子對于細胞的抗氧化應激反應至關重要,它的活性受到泛素化機制的調控。既往報道,個別與泛素化調控機制相關的基因如KLHL7和TOPORS基因與IRD的致病機制有關[22-23]。Plafker等[21]利用酵母菌雙雜合系統發現RCBTB1蛋白很可能是CUL3-泛素E3連接酶復合物的組成部分。研究報道,2例RCBTB1基因突變患者的外周血單核細胞中NFE2L2通路相關的基因表達水平顯著降低[5]。健康對照來源的iPSC-RPE細胞中證實了RCBTB1蛋白與CUL3-泛素E3連接酶復合物存在相互作用[15]。研究報道,在RCBTB1基因突變患者來源的iPSC-RPE細胞中檢測到與NFE2L2通路相關的基因表達水平較對照組顯著降低。
研究表明,RPE細胞可以通過激活NFE2L2通路增強抗氧化防御以減少由活性氧(ROS)引起的細胞損害[24-25]。筆者團隊近來在患者來源iPSC-RPE中發現即使在低濃度過氧叔丁醇(tBHP),一種常用的氧化劑,刺激下也產生大量ROS,表明RCBTB1基因缺陷的RPE細胞比對照組更容易受到tBHP誘導的氧化應激的影響[15],這一發現與先前的研究結果一致[14]。tBHP處理后的對照RPE細胞中RCBTB1蛋白表達水平顯著上調,這表明RCBTB1在RPE細胞氧化應激響應中起著一定作用。鑒于NFE2L2結合的底物適配蛋白Kelch樣環氧氯丙烷相關蛋白1(Keap1)是一種BTB-Kelch蛋白,通過與CUL3-泛素E3連接酶復合物結合來發揮功能,猜測底物適配蛋白RCBTB1和Keap1之間在CUL3-泛素E3連接酶復合物上的競爭解釋RCBTB1在調控NFE2L2水平中的作用。此外,NFE2L2轉錄因子通過結合其下游靶基因啟動子中的抗氧化應答元素(ARE)序列(TGACXXXGC)啟動抗氧化機制,而在RCBTB1啟動子位于轉錄起始位點上游33個核苷酸處存在一個ARE序列。綜上表明,在氧化應激條件下,NFE2L2可能作為正反饋回路的一部分,通過激活RCBTB1基因表達來發揮作用(圖2)。
圖2
RCBTB1基因突變可能致病機制示意圖
Cul3:Cullin3蛋白;E2:泛素連接酶;UBE2E3:泛素結合酶;Keap1:Kelch樣環氧氯丙烷相關蛋白1;Nrf2:核因子紅細胞衍生2樣;Ub:泛素;ROS:活性氧;TSS:轉錄起始位點;ARE:抗氧化應答元素;sMafs:小Maf蛋白
5 治療策略
針對早期發病表現為RP的RCBTB1基因相關遺傳性視網膜病變患者,基因治療應作為首選治療手段。早期病變患者仍保留部分尚未凋亡的RPE細胞及光感受器細胞,通過腺相關病毒(AAV)介導在視網膜下腔或玻璃體中導入攜帶有正常RCBTB1基因片段以替代突變的基因片段,理論上可以修復因RCBTB1蛋白表達不足或者缺失引起的視網膜功能障礙,改善患者視功能。由于在分子遺傳學領域取得的快速進展以及優化的基因傳遞載體的轉導效率,基因治療已經成為個性化治療遺傳性視網膜病變的重要手段。AAV在iPSC-RPE體外轉導過程和RPE的體內轉導表現出相似的轉導效率,這提示iPSC-RPE這一體外細胞模型在評估體內基因傳遞效力時具備實用性[26-27]。含有RCBTB1基因互補DNA(轉錄變體1,NM_018191)的AAV2/2和AAV2/8載體可用于高效轉導iPSC-RPE,并且AAV治療可實現RCBTB1基因互補DNA的長期活化水平表達,支持AAV基因療法在未來臨床試驗中提供持久療效的潛力[16]。然而考慮到RCBTB1高度表達可能會導致RPE細胞中NFE2L2激活通路的敏感性增高,這一點應在未來進行臨床基因治療中進行充分評估。
針對晚期發病表現為漸進性遲發型脈絡膜視網膜萎縮特征的患者,細胞替代治療更為合理,通過移植健康的iPSC-RPE移植片或者iPSC-RPE細胞懸浮液以替代原有的攜帶有RCBTB1基因突變的功能損害的RPE細胞,具備理論可行性。至于RPE細胞的純化、篩選以及如何將RPE移植片或懸浮液有效導入視網膜組織這些問題仍有待解決,需要研究學者與臨床工作者共同努力。多能干細胞移植的不同策略,包括單純的細胞移植、細胞外囊泡和細胞因子釋放,為未來干細胞移植研究方向提供了思路,包括改進細胞移植策略、提高細胞分化效率、解決免疫排斥問題以及推動治療方法的臨床應用[28]。
盡管基因替代RCBTB1基因治療的效果及將iPSC誘導分化為視網膜細胞的穩定與否仍然需要進一步研究,就目前而言,基因療法及干細胞療法仍是治療RCBTB1基因相關遺傳性視網膜病變這一類由明確基因突變引起的遺傳性眼病的主流手段。既往研究表明,N-乙酰半胱氨酸抗氧化劑,可以降低氧化應激刺激條件下RPE細胞中約16%的ROS自由基水平,這提示抗氧化劑在治療RCBTB1基因相關的視網膜病變中可能起到一定療效[15]。
6 小結與展望
本研究歸納總結了來自不同種族的RCBTB1基因相關遺傳性視網膜病變患者的眼部特征及遺傳學特征,提示RCBTB1基因相關視網膜病變的遺傳方式為常染色體隱性遺傳。基于既往研究成果對RCBTB1基因在視網膜細胞中的功能影響及治療策略展開分析,推測其在維持RPE細胞線粒體功能及抗氧化應激防御機制中發揮重要作用,因而應用基因替代治療及干細胞療法治療RCBTB1基因相關遺傳性視網膜病變具備可行性。本研究為探索此類罕見遺傳性視網膜病變的發病機制提供研究思路,為RCBTB1的基因治療及干細胞相關研究提供重要參考,亦為RCBTB1基因治療盡早進入臨床試驗奠定基礎。未來為確定在RCBTB1相關視網膜病變的發病年齡或多器官受累是否存在基因型與表型的相關性,仍需收集更多攜帶RCBTB1基因突變的IRD病例,通過進一步的研究闡明RCBTB1基因在視網膜中的作用,如通過檢測RCBTB1缺陷的視網膜細胞中線粒體DNA拷貝數及抗氧化系統組分(如超氧化物歧化酶2、超氧化物歧化酶1和過氧化氫酶)的表達水平,以研究RCBTB1基因與線粒體的潛在關聯,更好地了解RCBTB1基因在氧化應激應答中的作用。此外,還需要評估AAV介導的RCBTB1基因替代療法在動物模型中的安全性和有效性,以期發現基因替代治療及干細胞療法治療的可行性。