

引用本文: 胡玉章. Wagner綜合征一家系臨床分析. 中華眼底病雜志, 2024, 40(10): 766-771. doi: 10.3760/cma.j.cn511434-20240117-00035 復制
版權信息: ?四川大學華西醫院華西期刊社《中華眼底病雜志》版權所有,未經授權不得轉載、改編
Wagner綜合征(WS)是一種罕見的遺傳性玻璃體視網膜疾病,其主要臨床特征包括玻璃體空腔以及玻璃體腔內的條帶狀、膜狀或紗網狀增生和牽引。WS可能導致視網膜脫離、脈絡膜視網膜萎縮、進行性夜盲、近視及早發性白內障[1]。目前的研究已證實,至少有9種VCAN基因的顯性遺傳變異與WS相關[2]。盡管國外對此病已有較多報道[2-4],但國內的相關文獻較為有限,僅有一個漢族家系的報告[1]。本研究對臨床擬診并經基因檢測確診的一個彝族WS家系進行了觀察,現總結其觀察結果報道如下。
1 對象和方法
本研究遵循《赫爾辛基宣言》原則,經成都愛迪眼科醫院醫學倫理委員會批準(批文號: 2023-L12-20)。所有受檢者均充分了解本研究目的并自愿簽署書面知情同意書。
2023年6月于成都愛迪眼科醫院臨床檢查擬診并經基因檢測確診的彝族WS一家系中4例患者和1名家系成員(圖1)納入本研究。其中,男性2名,女性3名。父母系近親結婚。
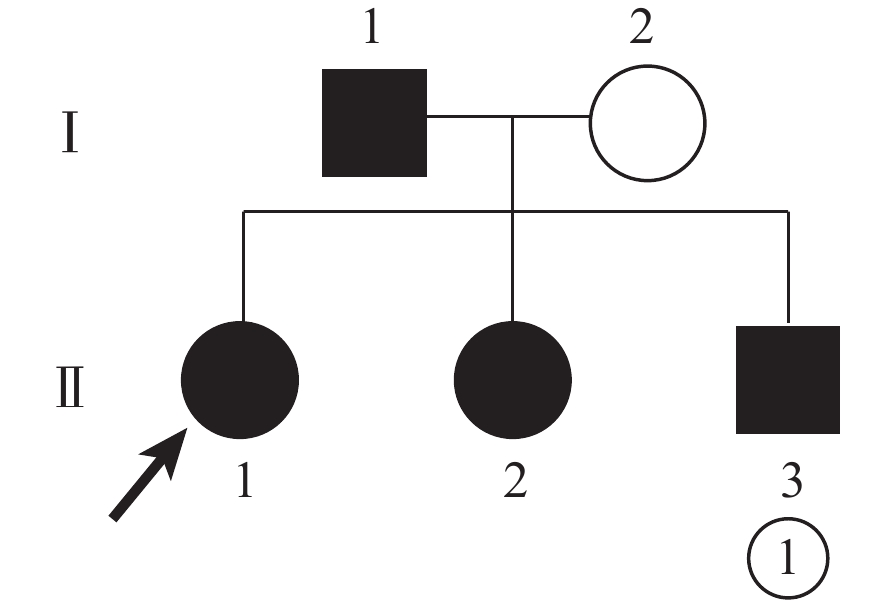 圖1
患者家系圖 ○:正常女性;●:女性患者;■:男性患者; ↗:先證者
圖1
患者家系圖 ○:正常女性;●:女性患者;■:男性患者; ↗:先證者
詳細詢問先證者病史,并行最佳矯正視力(BCVA)、眼壓、裂隙燈顯微鏡、間接檢眼鏡、眼底彩色照相、B型超聲檢查。先證者及其弟弟同時行眼底自發熒光(FAF)、光相干斷層掃描(OCT)、OCT血管成像檢查;全視野視網膜電圖(ERG)檢查1例(先證者)。先證者及其妹妹、弟弟同時行血糖、血壓、聽力、面部、關節、運動和全身體格檢查。
基因檢測及分子遺傳學分析均由北京智德醫學檢驗所完成。采集先證者及其家系成員2 ml外周靜脈血提取DNA。采用IDT xGen? Exome Research Panel v1.0和IDT xGen? Human mtDNA Research Panel對全外顯子組以及線粒體基因組進行捕獲,經Illumina NovaSeq 6000測序儀(PE150)行高通量測序,所得原始數據經過生物信息學分析處理后根據患者表型篩選出目的基因變異位點,對發現的致病變異位點進行Sanger測序檢驗。對數據的分析解讀參考美國醫學遺傳學和基因組學學院(ACMG)相關指南對變異位點進行致病性分析[5-7]。
2 結果
先證者(Ⅱ-1),女,23歲。自幼雙眼視力較差;無夜盲史。小學開始戴鏡,近視屈光度-4.00~-6.00 D,矯正欠佳。外院診斷為“雙眼先天性近視”。眼部檢查:右眼視力0.02,BCVA -8.00 D→0.1;左眼視力0.01,BCVA -8.00 D→0.1。右眼、左眼眼壓分別為8、9 mm Hg(1 mm Hg=0.133 kPa)。雙眼角膜透明;前房清晰。全視野ERG檢查,雙眼各波形態基本正常,部分振幅降低(圖2)。先證者妹妹(Ⅱ-2),20歲。雙眼視力0.6,BCVA 1.0;眼壓正常;眼底檢查未見明顯異常。先證者弟弟(Ⅱ-3),19歲。自幼雙眼視力差。2年前左眼因視網膜脫離、玻璃體嚴重增生在我院行玻璃體切割手術(PPV)聯合硅油填充治療;硅油取出手術后視網膜再脫離,再次行硅油填充,目前為硅油填充狀態。此次測得左眼BCVA光感,眼壓13 mm Hg;并發性白內障,眼底窺不清。5個月前,右眼曾行白內障手術,手術后矯正視力0.7;此次測得右眼視力0.02,-14.00 D→0.1;眼壓12 mm Hg。
 圖2
Wagner綜合征家系先證者(Ⅱ-1)雙眼全視野視網膜電圖像 各波形態基本正常,振幅降低
圖2
Wagner綜合征家系先證者(Ⅱ-1)雙眼全視野視網膜電圖像 各波形態基本正常,振幅降低
先證者及其妹妹(Ⅱ-2)、弟弟(Ⅱ-3)晶狀體均呈點狀、楔狀混濁,其弟弟更重;晶狀體后玻璃體空腔(圖3)。先證者雙眼、其弟弟右眼視網膜色澤較晦暗,鼻側、后極部團片狀暗區,周邊部對稱性視網膜面紗樣膜增生、牽拉,可見小片視網膜劈裂區;視網膜脈絡膜呈灶性和節段狀較大片對稱性萎縮;視盤傾斜(圖4);其弟弟視網膜血管呈毛刷狀向周邊部分布。FAF檢查,先證者及其弟弟視網膜后極部和鼻側均可見對稱性弱熒光區,表現相似(圖5A~5C)。OCT檢查,先證者及其弟弟均可見橢圓體帶部分缺失、斷裂(圖5D),脈絡膜層厚度降低(圖5E);先證者弟弟右眼視網膜神經上皮層明顯萎縮(圖5F)。OCTA檢查,先證者及其弟弟脈絡膜大中血管層萎縮、血流密度降低(圖5G~5I)。先證者及其妹妹、弟弟(Ⅰ-1~3)實驗室及全身體格檢查均未見明顯異常。
 圖3
Wagner綜合征家系先證者(Ⅱ-1)及其妹妹(Ⅱ-2)、弟弟(Ⅱ-3)右眼眼前節像 3A示先證者,晶狀體混濁;3B示先證者妹妹,晶狀體后玻璃體空腔;3C示先證者弟弟,晶狀體核、皮質混濁
圖3
Wagner綜合征家系先證者(Ⅱ-1)及其妹妹(Ⅱ-2)、弟弟(Ⅱ-3)右眼眼前節像 3A示先證者,晶狀體混濁;3B示先證者妹妹,晶狀體后玻璃體空腔;3C示先證者弟弟,晶狀體核、皮質混濁
 圖4
Wagner綜合征家系先證者(Ⅱ-1)雙眼及其弟弟(Ⅱ-3)右眼彩色眼底像 4A、4B分別示先證者右眼、左眼;4C示先證者弟弟右眼。視網膜色澤晦暗,周邊部玻璃體、視網膜面紗膜樣增生(紅箭),劈裂(白箭);視盤傾斜、血管走行呈“毛刷狀”(黃箭)
圖4
Wagner綜合征家系先證者(Ⅱ-1)雙眼及其弟弟(Ⅱ-3)右眼彩色眼底像 4A、4B分別示先證者右眼、左眼;4C示先證者弟弟右眼。視網膜色澤晦暗,周邊部玻璃體、視網膜面紗膜樣增生(紅箭),劈裂(白箭);視盤傾斜、血管走行呈“毛刷狀”(黃箭)
 圖5
Wagner綜合征家系先證者(Ⅱ-1)雙眼及其弟弟(Ⅱ-3)右眼眼底影像檢查像 5A~5C、5D~5F、5G~5I分別示先證者右眼、左眼,其弟弟右眼FAF、OCT、OCTA像。FAF檢查可見弱熒光區呈對稱分布,表現一致。OCT檢查可見橢圓體帶部分缺失、斷裂(紅箭);神經上皮層變薄(黃箭)、脈絡膜層明顯薄變(白箭)。OCTA檢查可見脈絡膜大中血管層萎縮,血流密度嚴重降低 FAF:眼底自發熒光;OCT:光相干斷層掃描;OCTA:OCT血管成像
圖5
Wagner綜合征家系先證者(Ⅱ-1)雙眼及其弟弟(Ⅱ-3)右眼眼底影像檢查像 5A~5C、5D~5F、5G~5I分別示先證者右眼、左眼,其弟弟右眼FAF、OCT、OCTA像。FAF檢查可見弱熒光區呈對稱分布,表現一致。OCT檢查可見橢圓體帶部分缺失、斷裂(紅箭);神經上皮層變薄(黃箭)、脈絡膜層明顯薄變(白箭)。OCTA檢查可見脈絡膜大中血管層萎縮,血流密度嚴重降低 FAF:眼底自發熒光;OCT:光相干斷層掃描;OCTA:OCT血管成像
基因檢測結果顯示,先證者及其父親(Ⅰ-1)、妹妹、弟弟攜帶VCAN基因c.9264A>G(p.Pro3088=)雜合變異。染色體位置chr5:82838 086,變異位點 c.9264A>G/p.Pro3088=(NM_004385.5)(Exon8)。該變異在人群基因頻率數據庫(gnomAD,https://gnomad.broadinstitute.org/)東亞人群、千人基因組計劃數據庫(http://www.1000genomes.org/)中國人群中均未檢測到。依據ACMG指南,該變異致病性為意義不明。先證者母親(Ⅱ-2)為野生型(圖6,7)。
 圖6
Wagner綜合征家系先證者(Ⅱ-1)高通量測序結果圖 VCAN基因c.9264A>G(p.Pro3088=)雜合變異(A:16次,G:21次)(紅箭)
圖6
Wagner綜合征家系先證者(Ⅱ-1)高通量測序結果圖 VCAN基因c.9264A>G(p.Pro3088=)雜合變異(A:16次,G:21次)(紅箭)
 圖7
Wagner綜合征家系先證者(Ⅱ-1)及其父母(Ⅰ-1、Ⅰ-2)、妹妹(Ⅱ-2)、弟弟(Ⅱ-3)基因測序圖 7A~7D分別示先證者及其妹妹、弟弟、父親,VCAN基因第8號外顯子的c.9264A>G雜合變異;7E示先證者母親,該位點不存在變異(紅箭)
圖7
Wagner綜合征家系先證者(Ⅱ-1)及其父母(Ⅰ-1、Ⅰ-2)、妹妹(Ⅱ-2)、弟弟(Ⅱ-3)基因測序圖 7A~7D分別示先證者及其妹妹、弟弟、父親,VCAN基因第8號外顯子的c.9264A>G雜合變異;7E示先證者母親,該位點不存在變異(紅箭)
3 討論
WS是一種少見的遺傳性常染色體顯性遺傳性玻璃體視網膜疾病,以玻璃體空腔和玻璃體腔內條帶狀、膜狀或紗網狀增生牽引為主要臨床表現,其可繼發視網膜脫離、脈絡膜視網膜萎縮、進行性夜盲、近視及早發白內障[1]。Wagner[8]于1938年首次在一個瑞士家庭的13名成員中發現WS并報道。WS具有近乎完全外顯率和可表達性的玻璃體視網膜病變,估計其患病率低于1/10×106[2-3, 9]。
WS最初特征性表現為裂隙燈顯微鏡下玻璃體光學空腔,伴周邊無血管玻璃體條索和紗樣膜、輕到高度近視,早期發生皮質性白內障和視網膜脈絡膜萎縮。隨病程進展可出現ERG振幅降低、視網膜劈裂和牽拉性視網膜脫離[3, 7, 10]。此外,還可見夜盲、視盤傾斜、中心凹異位、葡萄膜炎和青光眼等表現[9, 11-12]。
既往對WS的診斷均嚴格依據臨床特征和譜系分析。ERG檢查可見進行性廣泛視桿和視錐細胞功能障礙;視野呈環形暗點,中心視力進行性損失;FAF檢查可見周邊部和血管周圍進行性脈絡膜視網膜萎縮。這些特征也是色素性視網膜炎(RP)等其他幾種視網膜營養不良疾病的典型表現。基于這一表型重疊,遺傳分析已成為明確診斷的必要輔助手段[2, 10]。
文獻報道,55%的WS患者45歲前可能出現牽拉性視網膜脫離,87%的患者出現電生理異常[4]。由于WS臨床表型變化很大,容易導致誤診、漏診,如誤診為環形脈絡膜營養不良、Stickler綜合征(SS)等。本研究先證者弟弟左眼曾因視網膜全脫離行PPV治療,而今右眼出現周邊部玻璃體膜牽拉和視網膜劈裂。因此,基因檢測在WS診斷中發揮關鍵作用[11]。
本研究先證者及其弟弟發病較早,視力差且不能矯正;晶狀體點狀、楔狀和核混濁;晶狀體后玻璃體空腔;眼底改變一致,視網膜色澤晦暗伴色素沉著,玻璃體條索和視網膜面紗膜樣增生,與視網膜粘連,局限性視網膜牽拉和劈裂;視盤傾斜;視網膜脈絡膜呈灶性和節段狀較大片對稱性萎縮。OCT、OCTA檢查,先證者視網膜神經上皮層厚度無明顯改變,其弟弟神經上皮層明顯變薄;橢圓體帯斷裂、部分缺失,脈絡膜萎縮明顯變薄,特別是大中血管層,血流密度明顯降低。雙眼對稱性改變。其弟弟左眼視網膜全脫離行PPV治療。均符合WS臨床表現。最終經基因檢測發現VCAN基因變異,確定診斷。
先證者無夜盲史,全視野ERG檢查除振幅降低外,其余各波形態基本正常,可排除RP;先證者與其妹妹、弟弟無全身異常表現,如口腔、面部、聽力、運動和關節異常等,可與SS鑒別。Ⅰ型SS僅有眼部表現,無全身異常。但兩者在遺傳學方面存在明顯差異,通過基因檢測可進行鑒別[7]。患者均無進行性夜盲和向心性視野縮窄,可初步與侵蝕性玻璃體視網膜病變鑒別。
先證者與其妹妹、弟弟均無外傷和其他全身炎癥性疾病史。先證者及其弟弟眼底改變完全一致;其妹妹除晶狀體點狀混濁和玻璃體空腔外,視力和眼底未見明顯異常。由于患者晶狀體點狀混濁和視網膜劈裂區對視力無明顯影響,暫未給予更多治療,密切隨訪。有學者認為,視網膜脫離可發生在任一年齡段且可為雙側,建議適當情況下行預防性視網膜激光光凝或冷凍治療[3, 10, 13];但也有學者認為應密切觀察,對癥治療[3]。5個月前,先證者弟弟曾行右眼白內障手術,手術后矯正視力0.7。這說明,及時的干預可以改善視力狀況。同時臨床也應重視定期監測和適時治療的重要性,以預防潛在并發癥的發生。
先證者以及其父親、妹妹、弟弟均攜帶VCAN基因c.9264A>G(p.Pro3088=)雜合變異,為同義變異。該變異在gnomAD東亞人群、千人基因組數據庫中國人群中均未檢測到。依據ACMG標準,該變異滿足2個證據項:(1)PM2_Supporting:gnomAD數據庫中正常對照人群中未發現的變異(或隱性遺傳病中極低頻位點);(2)PP3:多種統計方法預測該變異可對基因或基因產物造成有害影響,包括保守性預測、進化預測、剪接位點影響等。經綜合評定判定該變異的致病性為“意義不明”,從分子水平無法確定該變異的眼部致病性[5-6,14-15]。目前已證實至少9種VCAN基因顯性遺傳突變與WS相關[2]。本研究WS家系患者VCAN基因變異位點是否為新變異位點以及患者眼部病變是否由此所致,尚需更多分析研究。
本研究對家系中3名成員進行了眼部更詳細的檢查,包括晶狀體照相、眼底彩色照相、FAF、OCT、OCTA、全視野ERG檢查等,更為全面地展示了WS在眼部的各種表現。本家系患者均為彝族,父母系近親結婚。國內報道WS一家系為漢族[1]。其變異位點不一致,是否與此有關,還需更多的病例分析研究。
常染色體顯性遺傳是患者雙親中必有一方患病,若父母雙方均未患病,所有子女均不患病。基因檢測發現先證者父親基因為雜合型,母親為野生型。父親VCAN基因變異,其3個子女均發現基因變異位點,3例患者均存在眼前節異常;先證者與其弟弟眼底出現病變。符合常染色體顯性遺傳規律[1]。
SS與WS既往被認為是同一疾病,稱為Wagner-Stickler綜合征。近年根據不同基因型和表型區分此兩種疾病。SS是一種常染色體顯性遺傳的進行性結締組織疾病,全身癥狀主要有口腔、面部、骨骼異常、身材矮小和關節表現;眼部可表現為高度近視、先天性大眼球、玻璃體異常和(或)視網膜脫離等。主要為Col2A、Col11A1、Col11A2基因變異。WS為先天性遺傳性玻璃體視網膜疾病,幾乎具有完全外顯率。突變基因主要是VCAN基因。眼部表現為近視、玻璃體空腔、視網膜格子樣變性和紗樣膜增生、脈絡膜視網膜萎縮、視網膜劈裂等;無全身異常表現[7, 15-17]。對于無全身異常僅有眼部表現的Ⅰ型SS,系Col2A基因變異所致[3, 16-17]。
Marfan綜合征、SS、WS是最常見與孔源性視網膜脫離(RRD)相關的遺傳性玻璃體視網膜病變綜合征。兒童RRD占所有RRD患者的0.5%~12.6%,因此應重視兒童和青少年RRD的致病原因是否與這些遺傳因素有關[4, 18]。隨著基因檢測技術普及和更為精準化,WS的患病率可能有所增加。
志謝 感謝成都愛迪眼科醫院的陳琛女士和周抒醫生在本研究的部分聯絡工作中所提供的幫助;感謝北京智德醫學檢驗所在遺傳基因檢測和解讀方面所做的貢獻
Wagner綜合征(WS)是一種罕見的遺傳性玻璃體視網膜疾病,其主要臨床特征包括玻璃體空腔以及玻璃體腔內的條帶狀、膜狀或紗網狀增生和牽引。WS可能導致視網膜脫離、脈絡膜視網膜萎縮、進行性夜盲、近視及早發性白內障[1]。目前的研究已證實,至少有9種VCAN基因的顯性遺傳變異與WS相關[2]。盡管國外對此病已有較多報道[2-4],但國內的相關文獻較為有限,僅有一個漢族家系的報告[1]。本研究對臨床擬診并經基因檢測確診的一個彝族WS家系進行了觀察,現總結其觀察結果報道如下。
1 對象和方法
本研究遵循《赫爾辛基宣言》原則,經成都愛迪眼科醫院醫學倫理委員會批準(批文號: 2023-L12-20)。所有受檢者均充分了解本研究目的并自愿簽署書面知情同意書。
2023年6月于成都愛迪眼科醫院臨床檢查擬診并經基因檢測確診的彝族WS一家系中4例患者和1名家系成員(圖1)納入本研究。其中,男性2名,女性3名。父母系近親結婚。
圖1
患者家系圖 ○:正常女性;●:女性患者;■:男性患者; ↗:先證者
詳細詢問先證者病史,并行最佳矯正視力(BCVA)、眼壓、裂隙燈顯微鏡、間接檢眼鏡、眼底彩色照相、B型超聲檢查。先證者及其弟弟同時行眼底自發熒光(FAF)、光相干斷層掃描(OCT)、OCT血管成像檢查;全視野視網膜電圖(ERG)檢查1例(先證者)。先證者及其妹妹、弟弟同時行血糖、血壓、聽力、面部、關節、運動和全身體格檢查。
基因檢測及分子遺傳學分析均由北京智德醫學檢驗所完成。采集先證者及其家系成員2 ml外周靜脈血提取DNA。采用IDT xGen? Exome Research Panel v1.0和IDT xGen? Human mtDNA Research Panel對全外顯子組以及線粒體基因組進行捕獲,經Illumina NovaSeq 6000測序儀(PE150)行高通量測序,所得原始數據經過生物信息學分析處理后根據患者表型篩選出目的基因變異位點,對發現的致病變異位點進行Sanger測序檢驗。對數據的分析解讀參考美國醫學遺傳學和基因組學學院(ACMG)相關指南對變異位點進行致病性分析[5-7]。
2 結果
先證者(Ⅱ-1),女,23歲。自幼雙眼視力較差;無夜盲史。小學開始戴鏡,近視屈光度-4.00~-6.00 D,矯正欠佳。外院診斷為“雙眼先天性近視”。眼部檢查:右眼視力0.02,BCVA -8.00 D→0.1;左眼視力0.01,BCVA -8.00 D→0.1。右眼、左眼眼壓分別為8、9 mm Hg(1 mm Hg=0.133 kPa)。雙眼角膜透明;前房清晰。全視野ERG檢查,雙眼各波形態基本正常,部分振幅降低(圖2)。先證者妹妹(Ⅱ-2),20歲。雙眼視力0.6,BCVA 1.0;眼壓正常;眼底檢查未見明顯異常。先證者弟弟(Ⅱ-3),19歲。自幼雙眼視力差。2年前左眼因視網膜脫離、玻璃體嚴重增生在我院行玻璃體切割手術(PPV)聯合硅油填充治療;硅油取出手術后視網膜再脫離,再次行硅油填充,目前為硅油填充狀態。此次測得左眼BCVA光感,眼壓13 mm Hg;并發性白內障,眼底窺不清。5個月前,右眼曾行白內障手術,手術后矯正視力0.7;此次測得右眼視力0.02,-14.00 D→0.1;眼壓12 mm Hg。
圖2
Wagner綜合征家系先證者(Ⅱ-1)雙眼全視野視網膜電圖像 各波形態基本正常,振幅降低
先證者及其妹妹(Ⅱ-2)、弟弟(Ⅱ-3)晶狀體均呈點狀、楔狀混濁,其弟弟更重;晶狀體后玻璃體空腔(圖3)。先證者雙眼、其弟弟右眼視網膜色澤較晦暗,鼻側、后極部團片狀暗區,周邊部對稱性視網膜面紗樣膜增生、牽拉,可見小片視網膜劈裂區;視網膜脈絡膜呈灶性和節段狀較大片對稱性萎縮;視盤傾斜(圖4);其弟弟視網膜血管呈毛刷狀向周邊部分布。FAF檢查,先證者及其弟弟視網膜后極部和鼻側均可見對稱性弱熒光區,表現相似(圖5A~5C)。OCT檢查,先證者及其弟弟均可見橢圓體帶部分缺失、斷裂(圖5D),脈絡膜層厚度降低(圖5E);先證者弟弟右眼視網膜神經上皮層明顯萎縮(圖5F)。OCTA檢查,先證者及其弟弟脈絡膜大中血管層萎縮、血流密度降低(圖5G~5I)。先證者及其妹妹、弟弟(Ⅰ-1~3)實驗室及全身體格檢查均未見明顯異常。
圖3
Wagner綜合征家系先證者(Ⅱ-1)及其妹妹(Ⅱ-2)、弟弟(Ⅱ-3)右眼眼前節像 3A示先證者,晶狀體混濁;3B示先證者妹妹,晶狀體后玻璃體空腔;3C示先證者弟弟,晶狀體核、皮質混濁
圖4
Wagner綜合征家系先證者(Ⅱ-1)雙眼及其弟弟(Ⅱ-3)右眼彩色眼底像 4A、4B分別示先證者右眼、左眼;4C示先證者弟弟右眼。視網膜色澤晦暗,周邊部玻璃體、視網膜面紗膜樣增生(紅箭),劈裂(白箭);視盤傾斜、血管走行呈“毛刷狀”(黃箭)
圖5
Wagner綜合征家系先證者(Ⅱ-1)雙眼及其弟弟(Ⅱ-3)右眼眼底影像檢查像 5A~5C、5D~5F、5G~5I分別示先證者右眼、左眼,其弟弟右眼FAF、OCT、OCTA像。FAF檢查可見弱熒光區呈對稱分布,表現一致。OCT檢查可見橢圓體帶部分缺失、斷裂(紅箭);神經上皮層變薄(黃箭)、脈絡膜層明顯薄變(白箭)。OCTA檢查可見脈絡膜大中血管層萎縮,血流密度嚴重降低 FAF:眼底自發熒光;OCT:光相干斷層掃描;OCTA:OCT血管成像
基因檢測結果顯示,先證者及其父親(Ⅰ-1)、妹妹、弟弟攜帶VCAN基因c.9264A>G(p.Pro3088=)雜合變異。染色體位置chr5:82838 086,變異位點 c.9264A>G/p.Pro3088=(NM_004385.5)(Exon8)。該變異在人群基因頻率數據庫(gnomAD,https://gnomad.broadinstitute.org/)東亞人群、千人基因組計劃數據庫(http://www.1000genomes.org/)中國人群中均未檢測到。依據ACMG指南,該變異致病性為意義不明。先證者母親(Ⅱ-2)為野生型(圖6,7)。
圖6
Wagner綜合征家系先證者(Ⅱ-1)高通量測序結果圖 VCAN基因c.9264A>G(p.Pro3088=)雜合變異(A:16次,G:21次)(紅箭)
圖7
Wagner綜合征家系先證者(Ⅱ-1)及其父母(Ⅰ-1、Ⅰ-2)、妹妹(Ⅱ-2)、弟弟(Ⅱ-3)基因測序圖 7A~7D分別示先證者及其妹妹、弟弟、父親,VCAN基因第8號外顯子的c.9264A>G雜合變異;7E示先證者母親,該位點不存在變異(紅箭)
3 討論
WS是一種少見的遺傳性常染色體顯性遺傳性玻璃體視網膜疾病,以玻璃體空腔和玻璃體腔內條帶狀、膜狀或紗網狀增生牽引為主要臨床表現,其可繼發視網膜脫離、脈絡膜視網膜萎縮、進行性夜盲、近視及早發白內障[1]。Wagner[8]于1938年首次在一個瑞士家庭的13名成員中發現WS并報道。WS具有近乎完全外顯率和可表達性的玻璃體視網膜病變,估計其患病率低于1/10×106[2-3, 9]。
WS最初特征性表現為裂隙燈顯微鏡下玻璃體光學空腔,伴周邊無血管玻璃體條索和紗樣膜、輕到高度近視,早期發生皮質性白內障和視網膜脈絡膜萎縮。隨病程進展可出現ERG振幅降低、視網膜劈裂和牽拉性視網膜脫離[3, 7, 10]。此外,還可見夜盲、視盤傾斜、中心凹異位、葡萄膜炎和青光眼等表現[9, 11-12]。
既往對WS的診斷均嚴格依據臨床特征和譜系分析。ERG檢查可見進行性廣泛視桿和視錐細胞功能障礙;視野呈環形暗點,中心視力進行性損失;FAF檢查可見周邊部和血管周圍進行性脈絡膜視網膜萎縮。這些特征也是色素性視網膜炎(RP)等其他幾種視網膜營養不良疾病的典型表現。基于這一表型重疊,遺傳分析已成為明確診斷的必要輔助手段[2, 10]。
文獻報道,55%的WS患者45歲前可能出現牽拉性視網膜脫離,87%的患者出現電生理異常[4]。由于WS臨床表型變化很大,容易導致誤診、漏診,如誤診為環形脈絡膜營養不良、Stickler綜合征(SS)等。本研究先證者弟弟左眼曾因視網膜全脫離行PPV治療,而今右眼出現周邊部玻璃體膜牽拉和視網膜劈裂。因此,基因檢測在WS診斷中發揮關鍵作用[11]。
本研究先證者及其弟弟發病較早,視力差且不能矯正;晶狀體點狀、楔狀和核混濁;晶狀體后玻璃體空腔;眼底改變一致,視網膜色澤晦暗伴色素沉著,玻璃體條索和視網膜面紗膜樣增生,與視網膜粘連,局限性視網膜牽拉和劈裂;視盤傾斜;視網膜脈絡膜呈灶性和節段狀較大片對稱性萎縮。OCT、OCTA檢查,先證者視網膜神經上皮層厚度無明顯改變,其弟弟神經上皮層明顯變薄;橢圓體帯斷裂、部分缺失,脈絡膜萎縮明顯變薄,特別是大中血管層,血流密度明顯降低。雙眼對稱性改變。其弟弟左眼視網膜全脫離行PPV治療。均符合WS臨床表現。最終經基因檢測發現VCAN基因變異,確定診斷。
先證者無夜盲史,全視野ERG檢查除振幅降低外,其余各波形態基本正常,可排除RP;先證者與其妹妹、弟弟無全身異常表現,如口腔、面部、聽力、運動和關節異常等,可與SS鑒別。Ⅰ型SS僅有眼部表現,無全身異常。但兩者在遺傳學方面存在明顯差異,通過基因檢測可進行鑒別[7]。患者均無進行性夜盲和向心性視野縮窄,可初步與侵蝕性玻璃體視網膜病變鑒別。
先證者與其妹妹、弟弟均無外傷和其他全身炎癥性疾病史。先證者及其弟弟眼底改變完全一致;其妹妹除晶狀體點狀混濁和玻璃體空腔外,視力和眼底未見明顯異常。由于患者晶狀體點狀混濁和視網膜劈裂區對視力無明顯影響,暫未給予更多治療,密切隨訪。有學者認為,視網膜脫離可發生在任一年齡段且可為雙側,建議適當情況下行預防性視網膜激光光凝或冷凍治療[3, 10, 13];但也有學者認為應密切觀察,對癥治療[3]。5個月前,先證者弟弟曾行右眼白內障手術,手術后矯正視力0.7。這說明,及時的干預可以改善視力狀況。同時臨床也應重視定期監測和適時治療的重要性,以預防潛在并發癥的發生。
先證者以及其父親、妹妹、弟弟均攜帶VCAN基因c.9264A>G(p.Pro3088=)雜合變異,為同義變異。該變異在gnomAD東亞人群、千人基因組數據庫中國人群中均未檢測到。依據ACMG標準,該變異滿足2個證據項:(1)PM2_Supporting:gnomAD數據庫中正常對照人群中未發現的變異(或隱性遺傳病中極低頻位點);(2)PP3:多種統計方法預測該變異可對基因或基因產物造成有害影響,包括保守性預測、進化預測、剪接位點影響等。經綜合評定判定該變異的致病性為“意義不明”,從分子水平無法確定該變異的眼部致病性[5-6,14-15]。目前已證實至少9種VCAN基因顯性遺傳突變與WS相關[2]。本研究WS家系患者VCAN基因變異位點是否為新變異位點以及患者眼部病變是否由此所致,尚需更多分析研究。
本研究對家系中3名成員進行了眼部更詳細的檢查,包括晶狀體照相、眼底彩色照相、FAF、OCT、OCTA、全視野ERG檢查等,更為全面地展示了WS在眼部的各種表現。本家系患者均為彝族,父母系近親結婚。國內報道WS一家系為漢族[1]。其變異位點不一致,是否與此有關,還需更多的病例分析研究。
常染色體顯性遺傳是患者雙親中必有一方患病,若父母雙方均未患病,所有子女均不患病。基因檢測發現先證者父親基因為雜合型,母親為野生型。父親VCAN基因變異,其3個子女均發現基因變異位點,3例患者均存在眼前節異常;先證者與其弟弟眼底出現病變。符合常染色體顯性遺傳規律[1]。
SS與WS既往被認為是同一疾病,稱為Wagner-Stickler綜合征。近年根據不同基因型和表型區分此兩種疾病。SS是一種常染色體顯性遺傳的進行性結締組織疾病,全身癥狀主要有口腔、面部、骨骼異常、身材矮小和關節表現;眼部可表現為高度近視、先天性大眼球、玻璃體異常和(或)視網膜脫離等。主要為Col2A、Col11A1、Col11A2基因變異。WS為先天性遺傳性玻璃體視網膜疾病,幾乎具有完全外顯率。突變基因主要是VCAN基因。眼部表現為近視、玻璃體空腔、視網膜格子樣變性和紗樣膜增生、脈絡膜視網膜萎縮、視網膜劈裂等;無全身異常表現[7, 15-17]。對于無全身異常僅有眼部表現的Ⅰ型SS,系Col2A基因變異所致[3, 16-17]。
Marfan綜合征、SS、WS是最常見與孔源性視網膜脫離(RRD)相關的遺傳性玻璃體視網膜病變綜合征。兒童RRD占所有RRD患者的0.5%~12.6%,因此應重視兒童和青少年RRD的致病原因是否與這些遺傳因素有關[4, 18]。隨著基因檢測技術普及和更為精準化,WS的患病率可能有所增加。
志謝 感謝成都愛迪眼科醫院的陳琛女士和周抒醫生在本研究的部分聯絡工作中所提供的幫助;感謝北京智德醫學檢驗所在遺傳基因檢測和解讀方面所做的貢獻






