

肺癌是全球最常見的惡性腫瘤之一,也是惡性腫瘤致死的首要原因,每年約200萬確診病例和176萬人因肺癌死亡[1]。氧化應激(oxidative stress,OS)被定義為氧化劑和抗氧化劑之間的失衡,導致氧化還原信號傳遞和控制的破壞以及分子損傷[2]。抗氧化能力異常被認為是腫瘤惡變的一個潛在機制[3]。多個OS基因參與肺癌的進展。AK4通過在缺氧狀態下增加細胞內活性氧(reactive oxygen species,ROS)水平來增加缺氧誘導因子-1α(hypoxia-inducible factor-1α,HIF-1α)蛋白表達,誘導上皮間充質轉化(epithelial mesenchymal transition,EMT),從而促進肺癌轉移[4]。KEAP1/NRF2通路被公認為是氧化還原平衡的關鍵調節因子,在肺癌中,NRF2的過度激活會導致其侵襲性和耐藥性的增加,以及對轉移的易感性[5]。此外,DNA甲基化通過調控ZDHHC1、NFE2L2及相關基因的表達來調節氧化還原穩態[6-7]。研究OS基因的潛在機制可能有助于確定肺癌的潛在致病因素和氧化還原相關的治療靶點。
孟德爾隨機化(Mendelian randomization,MR)是一種使用遺傳變異作為工具變量(instrumental variable,IVs)來探索暴露和結果之間潛在因果關系的方法。在MR中,使用等位基因的隨機分配避免了未觀察到的混雜因素(如生活方式和環境因素)帶來的偏差,以及反向因果關系的問題[8]。兩樣本MR分析可用于評估來自不同人群的工具變量與暴露和結局的關聯[9]。基于匯總數據的孟德爾隨機化(summary data-based Mendelian randomization,SMR)方法作為MR的延伸,將GWAS數據與基因表達、DNA甲基化進行整合,為鑒定基因表達或甲基化介導的因果效應提供了條件[10]。并采用HEIDI測試進行異質性檢驗,將潛在的因果關系與基因組中的廣泛連鎖不平衡(linkage disequilibrium,LD)區分開來[11]。本研究旨在通過SMR分析,探討OS基因與肺癌之間的潛在因果關系。
1 資料與方法
1.1 研究方法和數據來源
從GeneCards數據庫中獲得OS相關基因(
然后,從eQTLGen聯盟中獲取了OS基因在血液中表達量調控位點(eQTLs)的信息。eQTLGen聯盟是一個提供大規模血液eQTLs匯總統計數據的在線資源,包括來自37個數據集的31684例樣本基因表達的遺傳數據[15]。使用P<1×10-5作為顯著水平,篩選出與OS基因表達相關的eQTLs。
最后,從McRae等[16]提供的在線資源中獲取了OS基因在血液中DNA甲基化水平調控位點(mQTLs)的信息。該資源基于2個歐洲隊列的Meta分析:布里斯班系統遺傳學研究(n=614)和洛錫安出生隊列(n=1366)[17]。使用P<1×10-5作為顯著水平,篩選出與OS基因DNA甲基化水平相關的mQTLs。目前的研究關注順式-eQTLs和順式-mQTLs,即距離基因起始位點和終止位點1 Mb范圍內的單核苷酸多態性(single nucleotide polymorphisms,SNPs)[18]。
1.2 統計分析
本研究主要采用了SMR分析和共定位分析兩種方法,以探索OS基因與肺癌風險之間的因果關系。
作為MR的擴展,SMR被用來估計遺傳決定性狀(例如,基因表達、DNA甲基化作為暴露)和感興趣的性狀(例如疾病表型)之間的關聯。SMR分析的優勢是它可以同時利用GWAS和eQTL/mQTL數據,避免了樣本重疊和混雜因素的影響。使用PLINK軟件對GWAS SNPs、eQTLs和mQTLs進行質量控制,去除缺失率>0.05、未映射到人類參考基因組hg19的SNPs。使用SMR軟件進行了SMR分析和HEIDI檢驗,并對多重假設檢驗進行FDR校正。HEIDI檢驗是用來檢測異質性的一種方法,它的原理是比較每個SNP對暴露和結果的效應大小是否一致。如果一個SNP對暴露和結果的效應大小不一致,那么它可能是一個異質性SNP,也就是說它可能同時影響暴露和結果,但不是通過因果關系。HEIDI檢驗可以幫助我們篩選出那些可能存在混雜因素或者水平混雜的SNP,從而提高SMR分析的有效性和準確性。FDR校正是用來控制多重假設檢驗中錯誤發現率的一種方法,它的原理是根據P值大小進行排序和校正,從而得到每個假設被錯誤拒絕的概率。FDR校正可以幫助我們在保持較高統計功效的同時,避免過多的假陽性結果。
共定位分析是一種基于貝葉斯理論的方法,旨在研究不同性狀在給定區域內是否共享同一個因果變異。共定位分析可以幫助我們判斷兩個性狀是否有共同的遺傳機制,并為后續的功能驗證提供候選變異。我們使用coloc R軟件包進行了共定位分析,使用PPH4>0.8作為兩個性狀之間共享遺傳變異的閾值。PPH4是共定位分析中用來衡量兩個性狀是否共享同一個因果變異的概率的一種指標,它的原理是計算給定區域內存在一個共同因果變異的后驗概率。Giambartolomei等[19]認為PPH4>0.8作為閾值可以在保證較高靈敏度和特異度的同時,減少假陽性和假陰性結果。
2 結果
2.1 氧化應激相關基因的順式eQTLs與肺癌風險的SMR分析
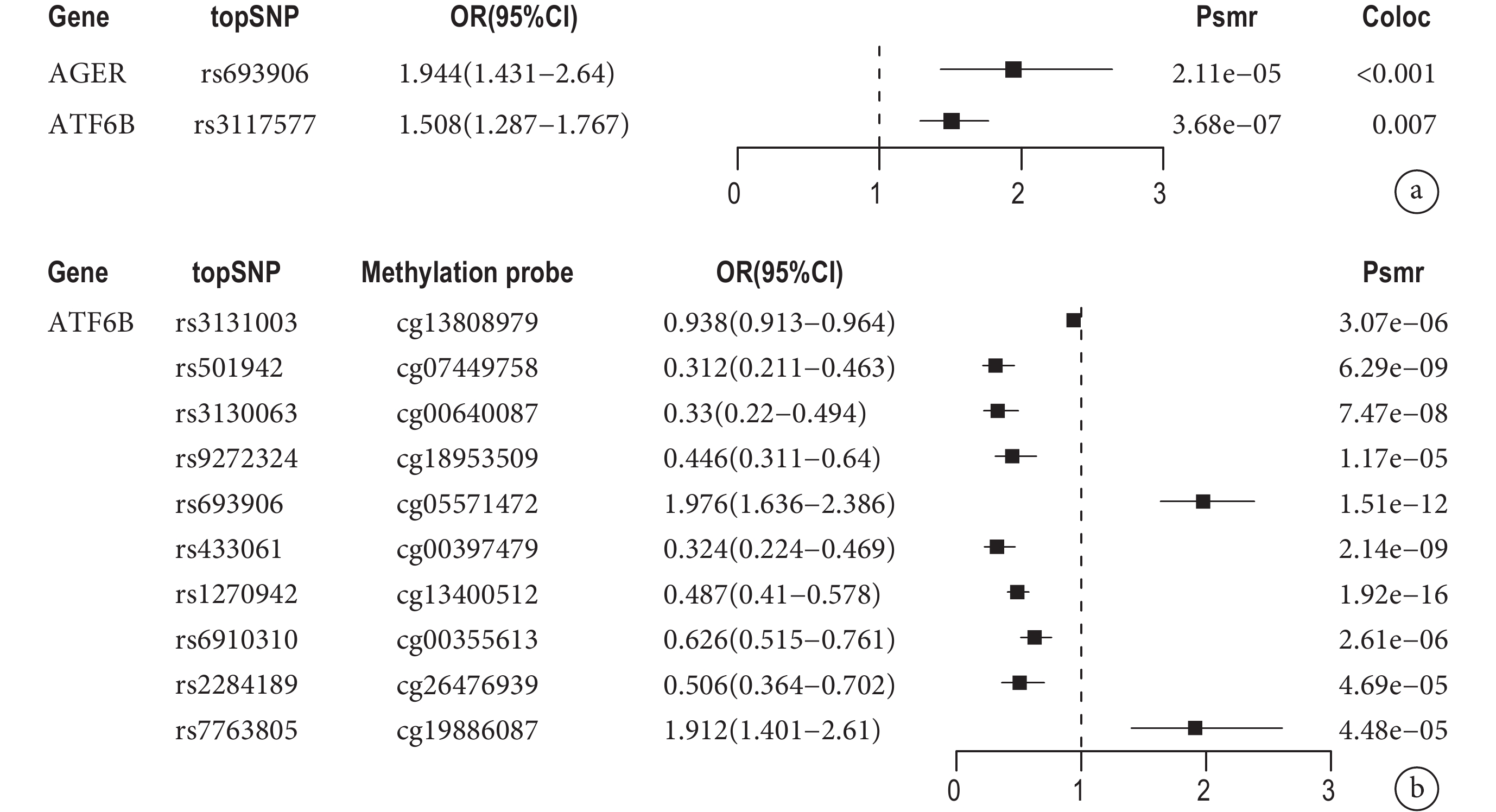
從GeneCards數據庫中獲得1188個與OS相關基因。首先通過SMR分析,得到與肺癌相關的工具變量。進行FDR校正(FDR<0.05),確定具有強相關性的SNP,然后進行HEIDI檢驗(PHEIDI>0.01),排除多效性的干擾。識別出2個基因與肺癌具有因果關系,進一步進行共定位分析,如果在基因表達和肺癌之間存在共同因果變異的后驗概率(PPH4)>0.8,則存在肺癌GWAS和eQTL共定位的證據。因果估計效應用OR值表示,AGER表達增加與肺癌風險增加相關[OR=1.944,95%CI(1.431,2.640),P<0.001],ATF6B表達增加也與肺癌風險增加相關[OR=1.508,95%CI(1.287,1.767),P<0.001](圖1a)。
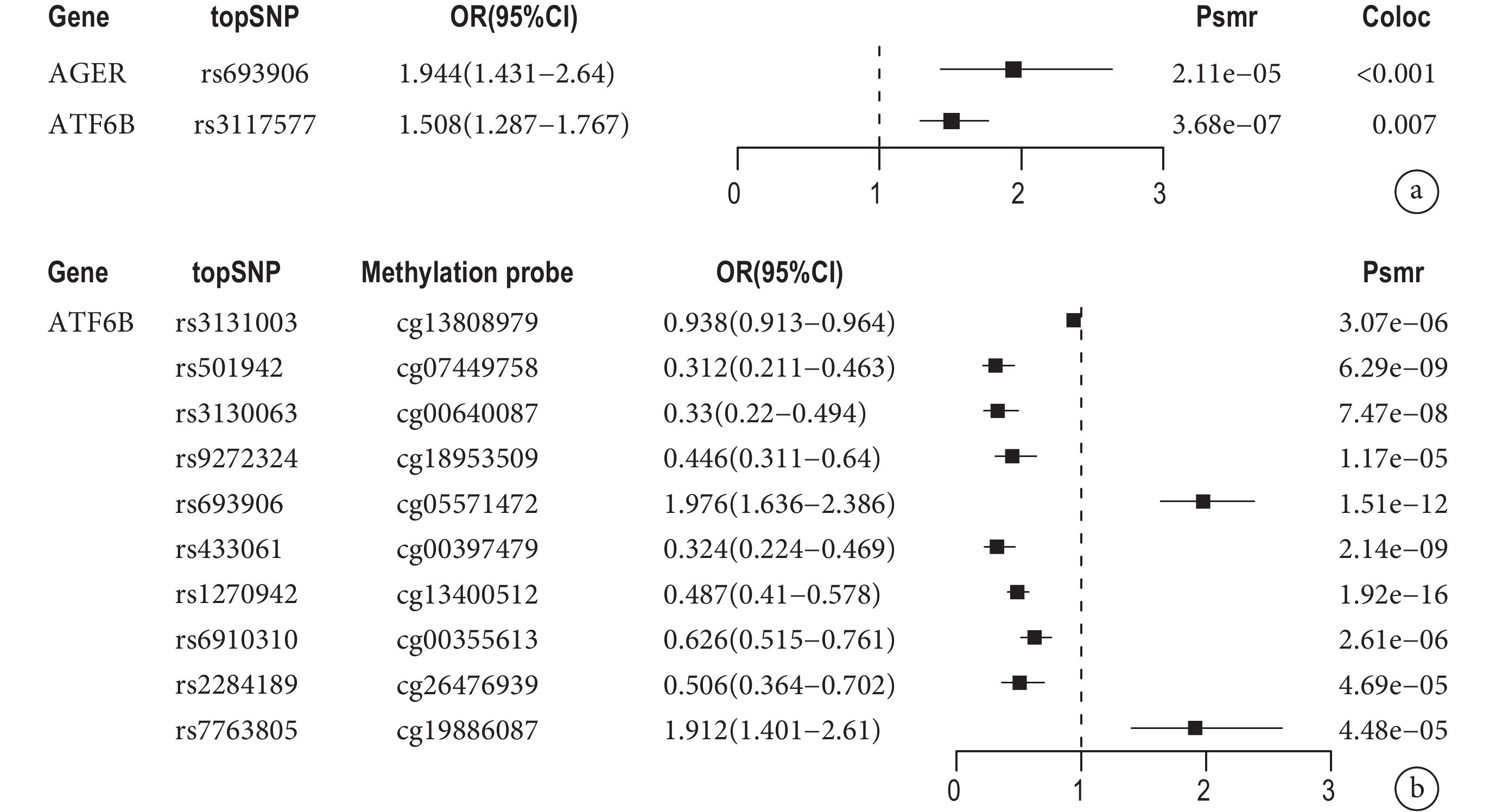 圖1
SMR分析結果
圖1
SMR分析結果
a:OS相關基因表達與肺癌風險之間的SMR分析結果;b:OS基因甲基化與肺癌風險之間的SMR分析結果;OS:氧化應激;SMR:基于匯總數據的孟德爾隨機化
2.2 氧化應激相關基因的順式mQTLs與肺癌風險的SMR分析
位于啟動子或增強子的DNA甲基化通常會影響疾病相關靶基因的調控。對于OS基因的DNA甲基化與肺癌預后之間的因果關系,進行SMR分析,同時進行FDR校正(FDR<0.05)和HEIDI檢驗(PHEIDI>0.05)。在ATF6B基因中發現10個甲基化位點與肺癌的預后相關(圖1b)。
3 討論
本研究探討了OS相關的遺傳決定因素對肺癌的潛在因果效應,并確定了AGER和ATF6B兩個與此有關的關鍵基因。這為連接遺傳位點、基因表達和甲基化與肺癌的潛在機制提供了初步證據。
AGER基因編碼一種細胞表面受體,它是免疫球蛋白超家族成員之一。此基因具有多種單核苷酸多態性(SNPs),這些變異可能會影響編碼蛋白的功能[20]。在乳腺癌和其他許多癌癥組織中,AGER的過表達已得到證實[21-22]。同時,AGER被視為促進癌癥惡病質特征的潛在分子決定因素,因此,以AGER為目標的治療策略可能具有對抗惡病質綜合征的潛力[23]。ATF6是一種受內質網(endoplasmic reticulum,ER)應激調控的轉錄因子,ATF6B是ATF6的一種亞型[24]。ATF6B基因的遺傳變異與乳腺癌的風險有關[25],并且與結直腸癌的風險也存在關聯[26]。在本研究中,我們發現ATF6B基因的表達與肺癌之間存在潛在的因果關系。
本研究的一個顯著優勢是,我們探討了所有OS相關基因與肺癌之間的因果關系,從而避免了之前研究中可能存在的選擇偏見。我們選擇了SMR作為主要的分析方法,并進行了共定位分析,這使我們的研究結果更為可靠。本研究也存在一些局限性。首先,由于當前分析方法和可用數據集的限制,我們只關注了OS基因的順式區域,未能考慮反式eQTL可能對調控網絡產生的廣泛影響[15]。未來研究中,探索反式eQTL的作用將是理解OS基因與肺癌關系的重要補充。其次,我們采用的貝葉斯共定位方法主要依賴于兩個性狀共享的單個因果變異。雖然這種方法被廣泛應用于多項研究中,并在探索基因表達和疾病關聯方面顯示出有效性[11,18]。但我們承認,當存在多個因果變異時,這種方法可能無法準確反映復雜的遺傳交互作用。Wallace等[27]在PLOS Genetics上發表的研究提出了一種考慮多因果變異的共定位分析方法。盡管我們當前的分析沒有采用這種方法,但我們認為這是未來研究的重要發展方向,并計劃在后續工作中探索這些新方法的應用。
綜上所述,盡管我們使用SMR分析發現AGER和ATF6B與肺癌存在潛在的因果關系,但這些發現仍然需要進一步驗證。SMR分析有其局限性。因此,我們的發現可以視為一個初步的探索,并不能作為確定性的證據。未來的研究可以進一步探索這些基因在肺癌發病機制中的具體作用,以及它們作為潛在藥物靶點的可能性。
利益沖突:無。
作者貢獻:周嗣丁負責論文設計,數據整理與分析,論文初稿撰寫等;肖洪璧、高銘駿負責數據整理與分析;汪夢夢負責論文校對和修改;王霄霖、束余聲負責論文設計、審閱與修改。
肺癌是全球最常見的惡性腫瘤之一,也是惡性腫瘤致死的首要原因,每年約200萬確診病例和176萬人因肺癌死亡[1]。氧化應激(oxidative stress,OS)被定義為氧化劑和抗氧化劑之間的失衡,導致氧化還原信號傳遞和控制的破壞以及分子損傷[2]。抗氧化能力異常被認為是腫瘤惡變的一個潛在機制[3]。多個OS基因參與肺癌的進展。AK4通過在缺氧狀態下增加細胞內活性氧(reactive oxygen species,ROS)水平來增加缺氧誘導因子-1α(hypoxia-inducible factor-1α,HIF-1α)蛋白表達,誘導上皮間充質轉化(epithelial mesenchymal transition,EMT),從而促進肺癌轉移[4]。KEAP1/NRF2通路被公認為是氧化還原平衡的關鍵調節因子,在肺癌中,NRF2的過度激活會導致其侵襲性和耐藥性的增加,以及對轉移的易感性[5]。此外,DNA甲基化通過調控ZDHHC1、NFE2L2及相關基因的表達來調節氧化還原穩態[6-7]。研究OS基因的潛在機制可能有助于確定肺癌的潛在致病因素和氧化還原相關的治療靶點。
孟德爾隨機化(Mendelian randomization,MR)是一種使用遺傳變異作為工具變量(instrumental variable,IVs)來探索暴露和結果之間潛在因果關系的方法。在MR中,使用等位基因的隨機分配避免了未觀察到的混雜因素(如生活方式和環境因素)帶來的偏差,以及反向因果關系的問題[8]。兩樣本MR分析可用于評估來自不同人群的工具變量與暴露和結局的關聯[9]。基于匯總數據的孟德爾隨機化(summary data-based Mendelian randomization,SMR)方法作為MR的延伸,將GWAS數據與基因表達、DNA甲基化進行整合,為鑒定基因表達或甲基化介導的因果效應提供了條件[10]。并采用HEIDI測試進行異質性檢驗,將潛在的因果關系與基因組中的廣泛連鎖不平衡(linkage disequilibrium,LD)區分開來[11]。本研究旨在通過SMR分析,探討OS基因與肺癌之間的潛在因果關系。
1 資料與方法
1.1 研究方法和數據來源
從GeneCards數據庫中獲得OS相關基因(
然后,從eQTLGen聯盟中獲取了OS基因在血液中表達量調控位點(eQTLs)的信息。eQTLGen聯盟是一個提供大規模血液eQTLs匯總統計數據的在線資源,包括來自37個數據集的31684例樣本基因表達的遺傳數據[15]。使用P<1×10-5作為顯著水平,篩選出與OS基因表達相關的eQTLs。
最后,從McRae等[16]提供的在線資源中獲取了OS基因在血液中DNA甲基化水平調控位點(mQTLs)的信息。該資源基于2個歐洲隊列的Meta分析:布里斯班系統遺傳學研究(n=614)和洛錫安出生隊列(n=1366)[17]。使用P<1×10-5作為顯著水平,篩選出與OS基因DNA甲基化水平相關的mQTLs。目前的研究關注順式-eQTLs和順式-mQTLs,即距離基因起始位點和終止位點1 Mb范圍內的單核苷酸多態性(single nucleotide polymorphisms,SNPs)[18]。
1.2 統計分析
本研究主要采用了SMR分析和共定位分析兩種方法,以探索OS基因與肺癌風險之間的因果關系。
作為MR的擴展,SMR被用來估計遺傳決定性狀(例如,基因表達、DNA甲基化作為暴露)和感興趣的性狀(例如疾病表型)之間的關聯。SMR分析的優勢是它可以同時利用GWAS和eQTL/mQTL數據,避免了樣本重疊和混雜因素的影響。使用PLINK軟件對GWAS SNPs、eQTLs和mQTLs進行質量控制,去除缺失率>0.05、未映射到人類參考基因組hg19的SNPs。使用SMR軟件進行了SMR分析和HEIDI檢驗,并對多重假設檢驗進行FDR校正。HEIDI檢驗是用來檢測異質性的一種方法,它的原理是比較每個SNP對暴露和結果的效應大小是否一致。如果一個SNP對暴露和結果的效應大小不一致,那么它可能是一個異質性SNP,也就是說它可能同時影響暴露和結果,但不是通過因果關系。HEIDI檢驗可以幫助我們篩選出那些可能存在混雜因素或者水平混雜的SNP,從而提高SMR分析的有效性和準確性。FDR校正是用來控制多重假設檢驗中錯誤發現率的一種方法,它的原理是根據P值大小進行排序和校正,從而得到每個假設被錯誤拒絕的概率。FDR校正可以幫助我們在保持較高統計功效的同時,避免過多的假陽性結果。
共定位分析是一種基于貝葉斯理論的方法,旨在研究不同性狀在給定區域內是否共享同一個因果變異。共定位分析可以幫助我們判斷兩個性狀是否有共同的遺傳機制,并為后續的功能驗證提供候選變異。我們使用coloc R軟件包進行了共定位分析,使用PPH4>0.8作為兩個性狀之間共享遺傳變異的閾值。PPH4是共定位分析中用來衡量兩個性狀是否共享同一個因果變異的概率的一種指標,它的原理是計算給定區域內存在一個共同因果變異的后驗概率。Giambartolomei等[19]認為PPH4>0.8作為閾值可以在保證較高靈敏度和特異度的同時,減少假陽性和假陰性結果。
2 結果
2.1 氧化應激相關基因的順式eQTLs與肺癌風險的SMR分析
從GeneCards數據庫中獲得1188個與OS相關基因。首先通過SMR分析,得到與肺癌相關的工具變量。進行FDR校正(FDR<0.05),確定具有強相關性的SNP,然后進行HEIDI檢驗(PHEIDI>0.01),排除多效性的干擾。識別出2個基因與肺癌具有因果關系,進一步進行共定位分析,如果在基因表達和肺癌之間存在共同因果變異的后驗概率(PPH4)>0.8,則存在肺癌GWAS和eQTL共定位的證據。因果估計效應用OR值表示,AGER表達增加與肺癌風險增加相關[OR=1.944,95%CI(1.431,2.640),P<0.001],ATF6B表達增加也與肺癌風險增加相關[OR=1.508,95%CI(1.287,1.767),P<0.001](圖1a)。
圖1
SMR分析結果
a:OS相關基因表達與肺癌風險之間的SMR分析結果;b:OS基因甲基化與肺癌風險之間的SMR分析結果;OS:氧化應激;SMR:基于匯總數據的孟德爾隨機化
2.2 氧化應激相關基因的順式mQTLs與肺癌風險的SMR分析
位于啟動子或增強子的DNA甲基化通常會影響疾病相關靶基因的調控。對于OS基因的DNA甲基化與肺癌預后之間的因果關系,進行SMR分析,同時進行FDR校正(FDR<0.05)和HEIDI檢驗(PHEIDI>0.05)。在ATF6B基因中發現10個甲基化位點與肺癌的預后相關(圖1b)。
3 討論
本研究探討了OS相關的遺傳決定因素對肺癌的潛在因果效應,并確定了AGER和ATF6B兩個與此有關的關鍵基因。這為連接遺傳位點、基因表達和甲基化與肺癌的潛在機制提供了初步證據。
AGER基因編碼一種細胞表面受體,它是免疫球蛋白超家族成員之一。此基因具有多種單核苷酸多態性(SNPs),這些變異可能會影響編碼蛋白的功能[20]。在乳腺癌和其他許多癌癥組織中,AGER的過表達已得到證實[21-22]。同時,AGER被視為促進癌癥惡病質特征的潛在分子決定因素,因此,以AGER為目標的治療策略可能具有對抗惡病質綜合征的潛力[23]。ATF6是一種受內質網(endoplasmic reticulum,ER)應激調控的轉錄因子,ATF6B是ATF6的一種亞型[24]。ATF6B基因的遺傳變異與乳腺癌的風險有關[25],并且與結直腸癌的風險也存在關聯[26]。在本研究中,我們發現ATF6B基因的表達與肺癌之間存在潛在的因果關系。
本研究的一個顯著優勢是,我們探討了所有OS相關基因與肺癌之間的因果關系,從而避免了之前研究中可能存在的選擇偏見。我們選擇了SMR作為主要的分析方法,并進行了共定位分析,這使我們的研究結果更為可靠。本研究也存在一些局限性。首先,由于當前分析方法和可用數據集的限制,我們只關注了OS基因的順式區域,未能考慮反式eQTL可能對調控網絡產生的廣泛影響[15]。未來研究中,探索反式eQTL的作用將是理解OS基因與肺癌關系的重要補充。其次,我們采用的貝葉斯共定位方法主要依賴于兩個性狀共享的單個因果變異。雖然這種方法被廣泛應用于多項研究中,并在探索基因表達和疾病關聯方面顯示出有效性[11,18]。但我們承認,當存在多個因果變異時,這種方法可能無法準確反映復雜的遺傳交互作用。Wallace等[27]在PLOS Genetics上發表的研究提出了一種考慮多因果變異的共定位分析方法。盡管我們當前的分析沒有采用這種方法,但我們認為這是未來研究的重要發展方向,并計劃在后續工作中探索這些新方法的應用。
綜上所述,盡管我們使用SMR分析發現AGER和ATF6B與肺癌存在潛在的因果關系,但這些發現仍然需要進一步驗證。SMR分析有其局限性。因此,我們的發現可以視為一個初步的探索,并不能作為確定性的證據。未來的研究可以進一步探索這些基因在肺癌發病機制中的具體作用,以及它們作為潛在藥物靶點的可能性。
利益沖突:無。
作者貢獻:周嗣丁負責論文設計,數據整理與分析,論文初稿撰寫等;肖洪璧、高銘駿負責數據整理與分析;汪夢夢負責論文校對和修改;王霄霖、束余聲負責論文設計、審閱與修改。