

4例女性Berry綜合征患兒,平均手術年齡為28.8 d,平均手術體重為3.64 kg,僅病例4是34 W+5早產兒,保守治療至71 d行手術治療,余3例均在新生兒期完成一期矯治手術,術后全部存活并隨訪至今。采用兩種不同的外科矯正技術用于修復主-肺動脈窗和異常起源的右肺動脈,包括1例采用了主動脈內擋板技術和另外3例進行了右肺動脈離斷再植術(其中1例原位重建,另2例右肺動脈前移到主動脈前方重建)。病例2使用主動脈內擋板術的患兒因術后右肺動脈狹窄經歷了兩次再次手術。病例3和病例4使用右肺動脈前移重建后的患兒隨訪右肺動脈發育均良好。Berry綜合征通過心臟彩色超聲和CT血管造影確診后,可以在有經驗的兒童心臟中心進行一期手術根治治療,安全有效。
Berry綜合征是一種極其罕見的復雜心血管畸形組合,1982年Berry首次進行了描述,其包括主-肺動脈窗(aortopulmonary window,APW)、右肺動脈異常起源于升主動脈(aortic origin of the right pulmonary artery,AORPA)、主動脈弓病變[包括主動脈弓中斷(interrupted aortic arch,IAA)、主動脈弓發育不良(hypoplastic aortic arch,HAA)、主動脈縮窄(coarctation of the aorta,CoA)],以及完整的室間隔[1]。Berry綜合征在先天性心臟病中的發生率約為0.046%,國內外文獻[2-3]有記載的病例不超過百例,通常表現出一種或多種臨床癥狀,如發紺、呼吸窘迫、心臟雜音、充血性心力衰竭等。大多數患者在出生后不久就死亡了,少數幸存者會出現嚴重的肺動脈高壓,因此早期、及時、準確的術前診斷和治療非常重要,而術前詳細評估該畸形的解剖特征對于制定手術策略非常關鍵。
基于麻醉、體外循環和重癥監護的日益發展,大多數中心提倡在新生兒期進行一期矯治手術[4-6]。本文通過回顧性分析浙江大學醫學院附屬兒童醫院收治的Berry綜合征患兒的臨床資料和手術策略,總結其解剖特征以及近遠期的隨訪結果,比較不同手術方式的優缺點,以期于優化這種罕見疾病的長遠療效。
臨床資料 4例患兒均通過經胸超聲心動圖和計算機斷層掃描血管成像(computerized tomographic angiography,CTA)明確診斷并根據其解剖特征進行如下分型:(1)根據Celoria和Patton的分類,確定IAA形態學診斷[7]:A型中斷發生在主動脈弓峽部,位于左鎖骨下動脈與動脈導管之間;B型中斷發生在左頸總動脈和左鎖骨下動脈之間;C型中斷發生在無名動脈和左頸總動脈之間。(2)APW類型是基于Mori的原始分類[8]:Ⅰ型是半月瓣緊上方的近端缺損;Ⅱ型是升主動脈更向頭部的遠端缺損;Ⅲ型是近端和遠端缺損相結合的完全缺損。(3)根據AORPA與APW的位置關系Berry將其分為2個亞型[1]:a型是指騎跨的右肺動脈(right pulmonary artery,RPA),血流動力學與升主動脈相關,但仍保持與主肺動脈和左肺動脈的解剖連續性;b型是另一種完全分離的RPA,在血流動力學和解剖學上都與升主動脈相關。
所有患兒均采用深低溫低流量順行選擇性腦灌注下進行主動脈弓重建。通過胸骨正中切口,插入2根動脈插管和上下腔插管建立體外循環:1根插入遠端升主動脈的右側,另1根插入動脈導管連接到降主動脈。轉流啟動后,左右肺動脈需立即阻斷,以防止肺循環充血。冷卻至合適的溫度后,阻斷升主動脈,將冷血心臟停搏液注入主動脈根部。然后拔除動脈導管插管,將升主動脈插管調整到無名動脈中,啟動深低溫低流量選擇性腦灌注。切斷縫扎動脈導管,充分游離升主動脈的分支及降主動脈,降主動脈上提后與主動脈弓部作端-側吻合,后壁直接吻合,前壁予以牛心包補片擴大成形。弓部修復完成后,將主動脈插管恢復到正常的弓位。然后通過主動脈前壁縱切口打開APW,進行徹底的檢查,分析APW的大小和位置、冠狀動脈的起源以及RPA的開口位置。
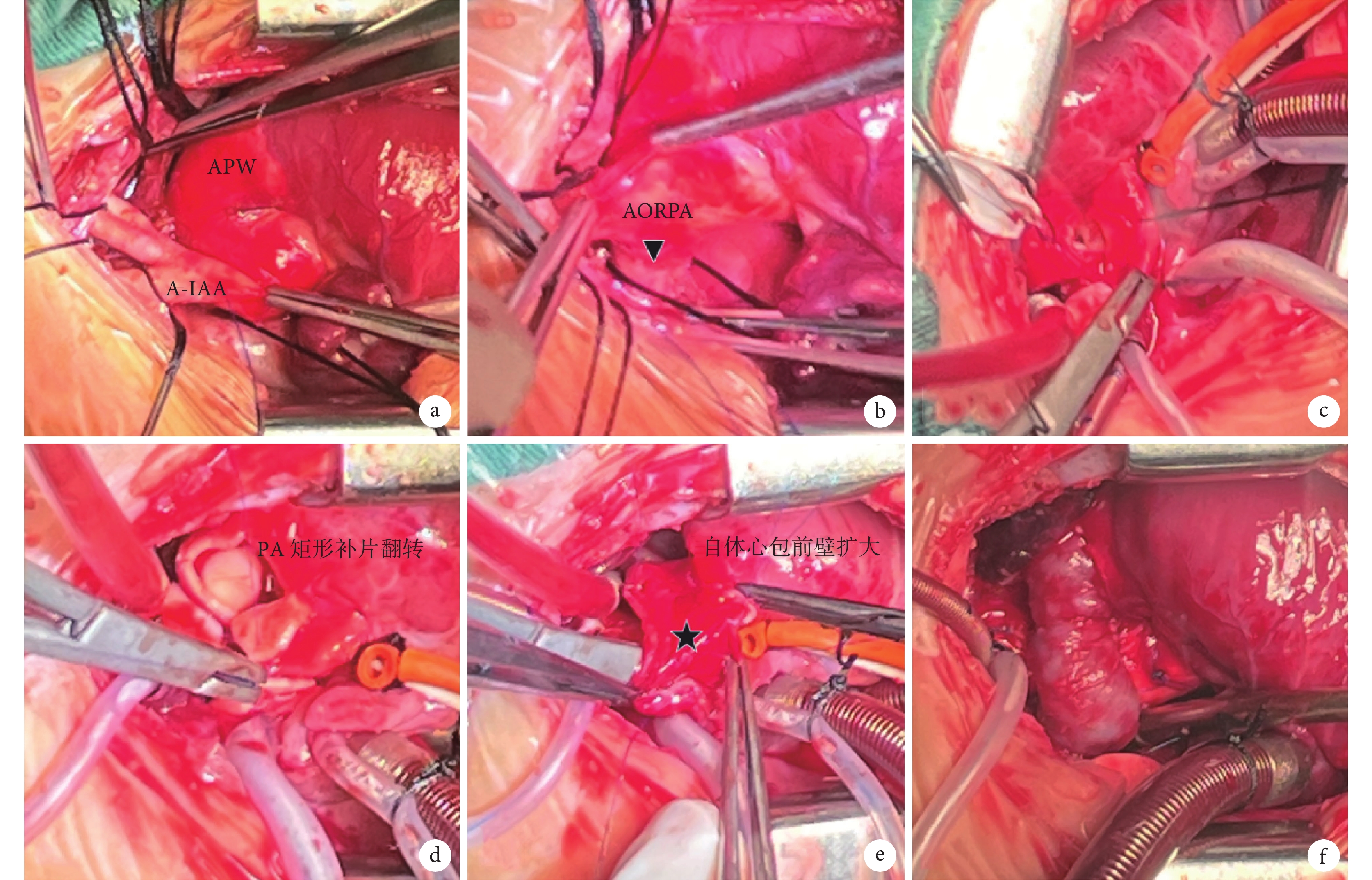
本中心采用了兩種手術方式來修復APW和AORPA:(1)主動脈內擋板技術:取合適大小的補片修補主動脈的缺損,并同時將RPA分隔至肺總動脈。(2)右肺動脈離斷再植技術:補片三明治法縫合修補APW,在主動脈后壁離斷RPA后用牛心包修補缺口,再將RPA移植到肺總動脈。本中心有3例患兒均使用了該手術方法,其中病例1的RPA在解剖原位移植到肺總動脈,病例3和4的RPA前移到主動脈的前方,肺總動脈的前壁取一矩形袢翻轉后與RPA后壁吻合,前壁用自體心包擴大成形。病例4術中的操作步驟見下圖1。
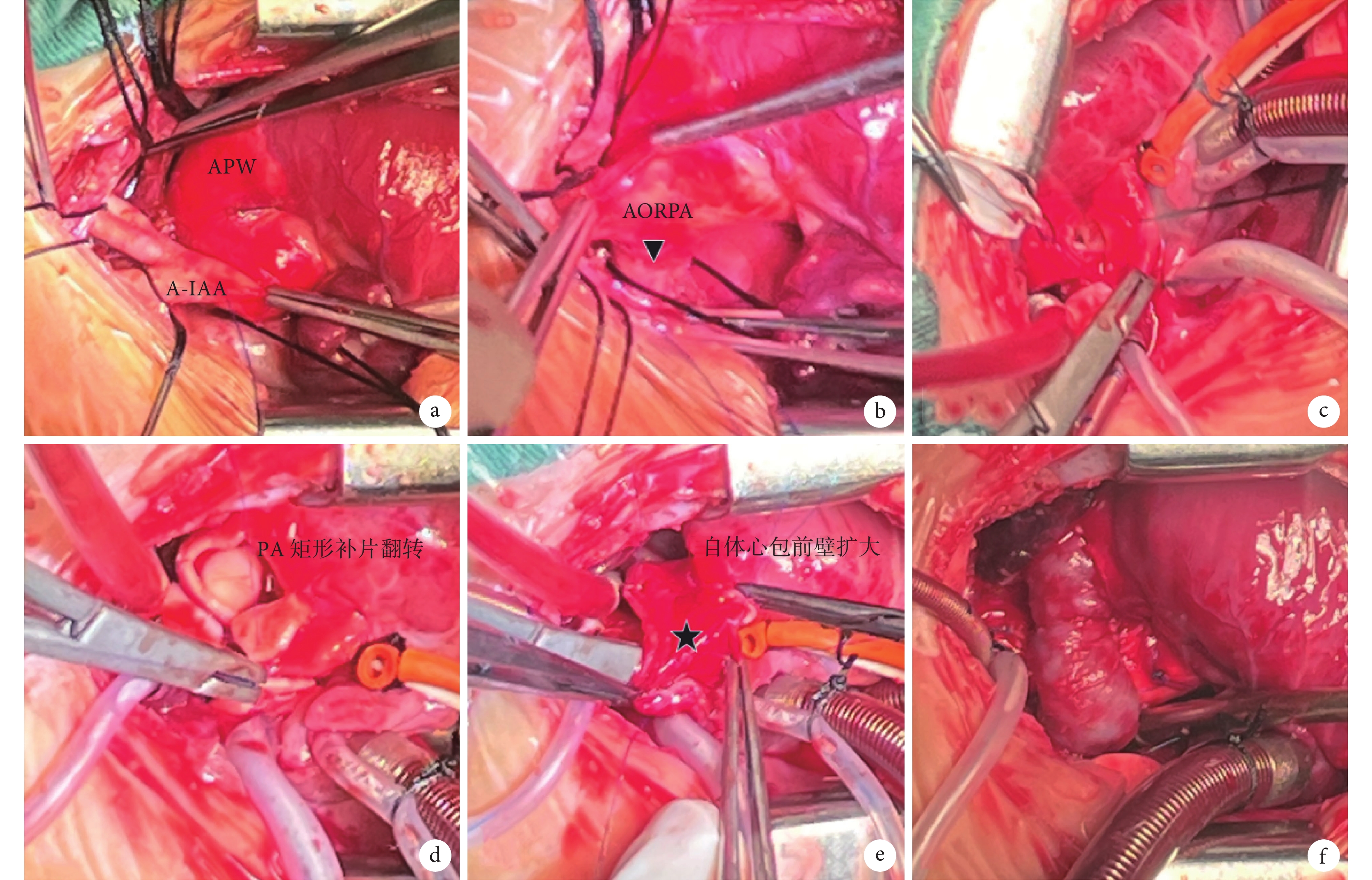 圖1
病例4 RPA前移重建的手術示意
圖1
病例4 RPA前移重建的手術示意
a:A型IAA,升主動脈發出三個分支后中斷;b:RPA從主動脈的后壁發出;c:采用補片三明治法縫合修補APW;d:肺總動脈的前壁取一矩形袢翻轉后與RPA后壁進行吻合;e:用自體心包擴大成形RPA的前壁;f:重建后趴在主動脈前方的RPA;IAA:主動脈弓中斷;APW:主-肺動脈窗;AORPA:右肺動脈起源于升主動脈;RPA:右肺動脈
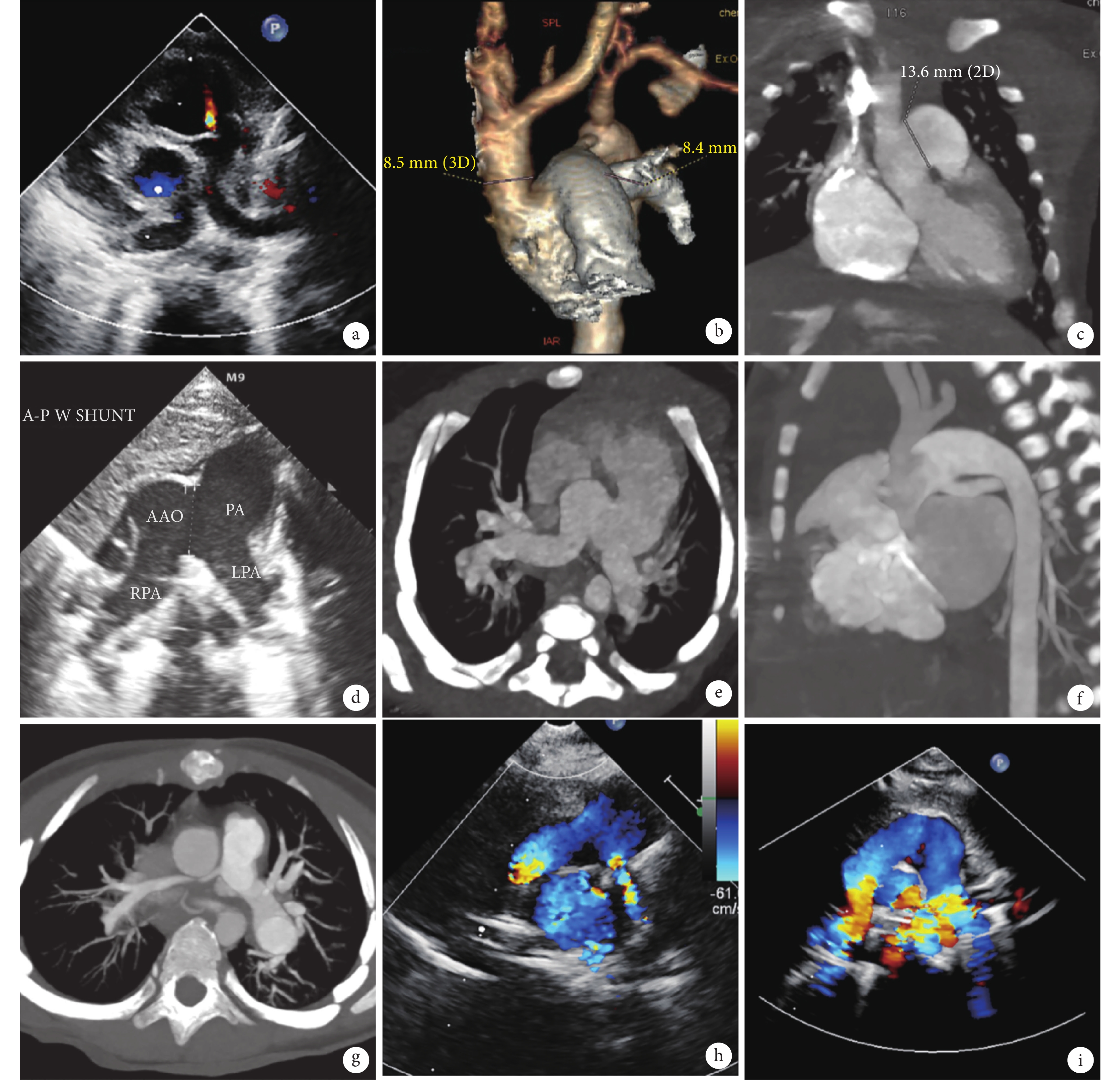
病例1,女,單胎40 W出生,因紫紺、咳嗽入院,診斷為:A型IAA,II型APW,AORPA與APW的位置形態為a型(圖2a)。手術年齡為28 d,手術體重為3.65 kg,主動脈阻斷時間為94 min,選擇性腦灌時間為50 min,術后ICU滯留時間10 d,術后呼吸機治療3 d。手術采用了RPA離斷再植技術(原位移植到肺總動脈)。術后隨訪至今已6年余,最后一次心超RPA壓力梯度為25 mm Hg(1 mm Hg=0.133 kPa),降主動脈流速為1.0 m/s。
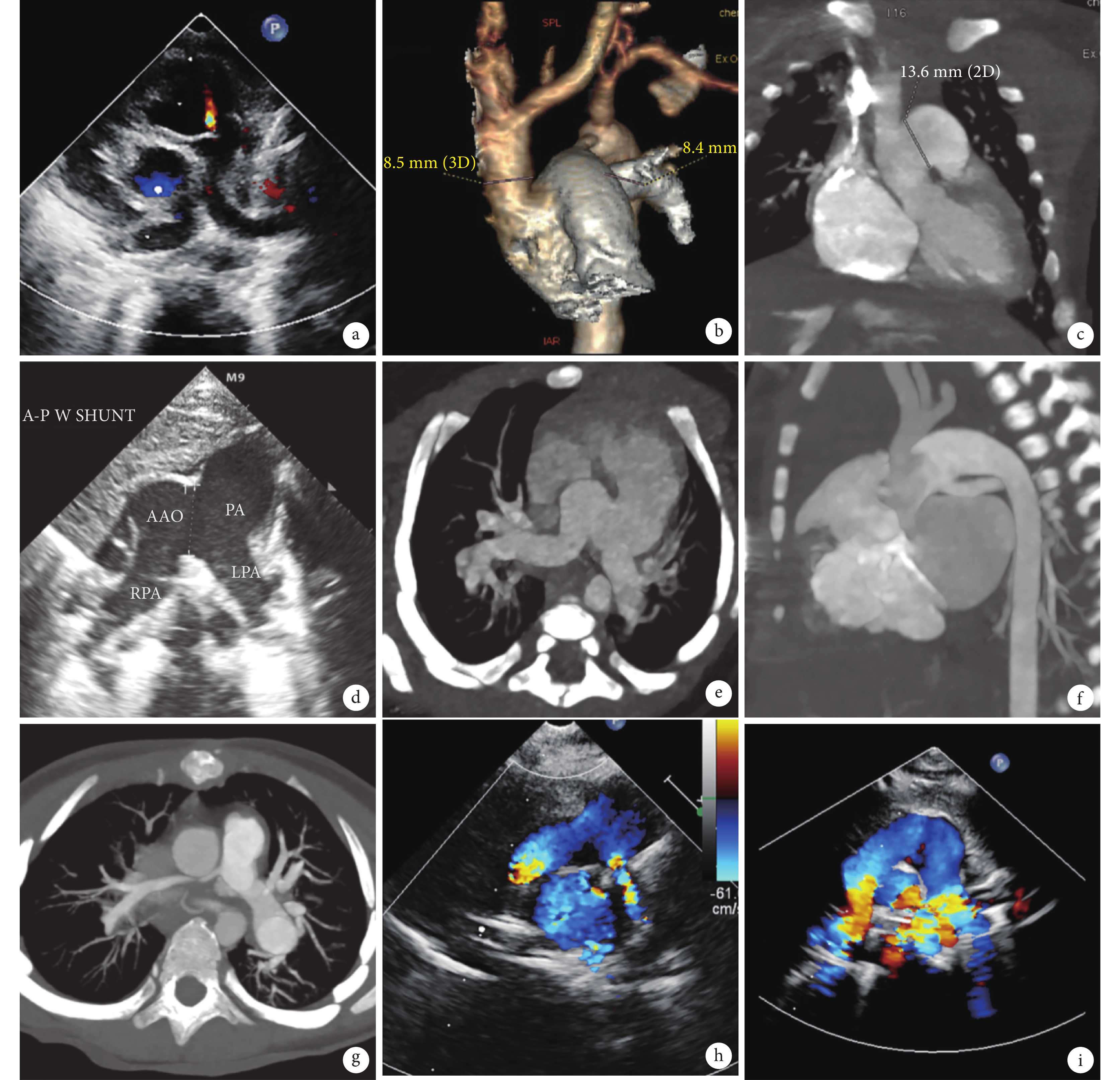 圖2
所有患兒的影像資料
圖2
所有患兒的影像資料
a:病例1的超聲切面可見特征性“蝴蝶征”影像;b:病例2的CTA三維成像顯示B型IAA;c:病例2的CT矢狀面成像顯示巨大的III型APW;d是病例3超聲切面的特征性“蝴蝶征”影像;e和f:病例4的CT成像顯示A型IAA、IIb型APW和AORPA;g:病例2術后隨訪時行CTA確診RPA再狹窄;h和f:分別是病例3和4術后超聲隨訪RPA發育良好無狹窄
病例2,女,因氣促、插管狀態下入院,診斷為:B型IAA,III型APW,AORPA與APW的位置形態為b型(圖2b、c)。手術年齡為8 d,手術體重為3.86 kg,主動脈阻斷時間為101 min,選擇性腦灌時間為79 min,術后呼吸機治療4 d,ICU期間發生了消化道大出血的并發癥,術后ICU滯留時間21 d,共住院50 d。手術采用了主動脈內擋板技術,肺總動脈前壁取一矩形袢并下翻用作主動脈內擋板的補片,然后用自體心包補片修補肺總動脈。然而患兒在隨訪期間發現RPA再狹窄(圖2g),術后定期常規超聲篩查提示RPA血流速度:2.7 m/s,壓差30 mm Hg,內徑0.3 cm,然后經CT血管造影(CTA)證實后,分別在初次手術后的第12個月和第22個月患兒接受了外科補片血管成形術。近期隨訪該患兒RPA和LPA都發育良好,最后一次心超RPA壓力梯度為9 mm Hg,降主動脈流速為1.6m/s。
病例3,女,單胎38 W+3出生,因黃疸入院,診斷為:A型IAA,II型APW,AORPA與APW的位置形態為b型(圖2d)。手術年齡為8 d,手術體重為3.05 kg,主動脈阻斷時間為108 min,選擇性腦灌時間為40 min,術后ICU滯留時間9 d,術后呼吸機治療2 d。手術采用了RPA離斷后前移重建技術。術后隨訪至今已2年余,最后一次心超RPA壓力梯度為3 mm Hg,降主動脈流速為1.3 m/s(圖2h)。
病例4,女,雙胎妊娠34 W+5早產兒,診斷為:A型IAA,II型APW,AORPA與APW的位置形態為b型(圖2e、f)。出生時體重僅2.30 kg,經過前列腺素E2開放動脈導管治療后,患兒生命體征平穩,遂繼續營養支持治療,待體重上升后再予以手術矯治,故其手術年齡為71 d,矯正胎齡為34 d,手術時體重為3.05 kg,主動脈阻斷時間為129 min,選擇性腦灌時間為34 min,術后呼吸機治療3 d,但因術后左側膈膨升經行膈肌折疊治療,術后ICU滯留時間為21 d,住院時間54 d。手術同樣采用了RPA離斷后前移重建技術。術后隨訪至今已半年余,最后一次心超RPA壓力梯度為4 mm Hg,降主動脈流速為1.5 m/s(圖2f)。
4例患者術后均順利出院并隨訪至今。病例3和病例4采用右肺動脈前移重建的方法,術后隨訪可見RPA發育佳,壓力梯度明顯較小。所有患兒均未出現喂養困難、聲帶麻痹、主動脈瓣上狹窄或主動脈弓再狹窄等其他并發癥。
本研究經浙江大學附屬兒童醫院倫理委員會批準(倫理批號:2023-IRB-0342-P-01),所有程序均符合機構和(或)國家研究委員會的倫理標準以及《赫爾辛基宣言》。
討論 Berry綜合征是一組極其罕見的先天性心血管畸形,其發病機制尚不明確,Berry等提出胚胎假說認為是APW影響了AORPA的形成,使流經主動脈弓的血流減少,從而導致HAA/CoA/IAA的發生[1]。由于其復雜性及危重性,如何在新生兒期給予正確的診斷和一期手術矯治仍然是臨床工作的難點。大多數患兒出生后早期即可出現嚴重的肺動脈高壓,APW和AORPA導致持續左向右分流,出現右肺動脈擴張,嚴重的甚至會壓迫氣道,而下半身的灌注則完全依賴于動脈導管[2]。多數學者建議在新生兒期對這些患者進行手術矯正,以保護肺血管床避免形成不可逆的肺高血,并盡早恢復下半身的正常血供[5-6,9]。根據Bi等[3]的Meta分析,大多數Berry綜合征為男性(49/73,67.1%),絕大部分可在產前或出生后3個月內明確診斷(56/81,69.1%),合并A型IAA(67/76,88.2%)、II型APW(56/71,78.9%)和b型AORPA為主(73/86,84.9%),未手術患者的死亡率為100%,死亡年齡的中位數是1個月。本中心的4例患兒除性別均是女性之外,其他臨床特征都與文獻描述相符,且均在新生兒期得到明確診斷,及全部完成一期手術矯治,術后存活率100%。
經胸超聲心動圖是出生后評估Berry綜合征的初始工具,這是一種安全且侵入性較小的方式,但由于有限的成像平面和視野,可能會出現AORPA的漏診,聯合CTA,出現RPA起源于升主動脈、左肺動脈起源于主肺動脈、APW位于主動脈和肺動脈之間,呈現“蝴蝶征”這種特征性影像時需高度警惕Berry綜合征的診斷[10]。本中心的4例患兒通過超聲聯合CTA,在新生兒期得到了明確診斷,并及時予以合適的治療策略。
手術修復的策略有兩種:(1)分期修復:第一階段放置肺動脈環縮帶控制肺血流,患兒通氣狀況改善后再接受完整的手術修復,Ghelani等[11-12]建議早產兒或小于胎齡兒應考慮分期修復。但隨著體外監護的日益發展,分期手術可酌情選擇。本中心病例4是早產兒且低體重,但經內科治療后患兒生命體征平穩,遂繼續營養支持治療,故其手術年齡為71 d,術后隨訪肺動脈壓力逐漸下降正常。(2)一期手術矯治:通過關閉APW,將RPA連接到肺總動脈,并建立主動脈弓連續性來實現的,大多數文獻報道一期矯治手術可獲得令人滿意的結果,且兩組之間術后1個月內的死亡率或1年隨訪期內的存活時間沒有顯著性差異[3,5,13]。矯治手術的解剖特點主要集中在APW的大小和位置,RPA異常起源的位置,以及IAA的類型和范圍,重建無張力且連續性的RPA和主動脈弓是影響手術結果的重要因素。本中心4例患兒均采用主動脈后壁端側吻合、前壁人工材料擴大成形修復主動脈弓,促進潛在的血管生長,術后均無主動脈弓再狹窄,無持續性肺動脈高壓,總手術風險可能類似于單純性IAA,近期和中期血流動力學和臨床結果均令人滿意。
本中心采用了2種手術方法重建RPA的連續性:(1)主動脈內擋板技術,技術要點是精確修整主動脈內補片的大小,防止左心室流出道和RPA的梗阻。胡等提出所有Ⅱa型APW和AORPA的Berry綜合征患兒均可采用該方法,但僅適用于部分Ⅱb型,這取決于APW到RPA的距離[4]。然而本中心的病例2(B型IAA和Ⅲ型APW)采用了該手術方法,術后出現了嚴重的RPA起點處的狹窄,說明該技術可能不適合Ⅲ型APW,考慮其補片較大,升主動脈血壓高,補片會被推向肺動脈導致壓迫狹窄。(2)RPA離斷再植技術,是孤立性RPA起源異常患兒最常采用的手術方法,可用于任何類型的Berry綜合征,無論RPA的位置或APW的大小。本中心其余3例均使用了該手術方法,其中病例1的RPA解剖原位移植到肺總動脈,病例3和4的RPA前移到主動脈的前方,與LeCompte操作相似,肺總動脈的前壁取一矩形袢翻轉后與RPA后壁吻合,前壁用自體心包擴大成形。與主動脈內擋板技術相比較,該方法破壞了RPA和MPA之間的原有連續性,且手術操作更復雜[14-16]。RPA前移重建的手術方法在個別病例報道中也獲得了良好的術后效果,Chang等[17]對1例伴有APW、AORPA、IAA和室間隔缺損的足月女嬰采用了RPA前移重建的一期手術矯治,術后氣道壓迫立即完全緩解,術后隨訪35個月CTA證實RPA發育良好。本中心2例采用RPA前移重建的患兒比1例采用RPA原位重建的患兒,獲得了更好的RPA發育,壓力梯度更小。
術后并發癥早期是院內死亡,遠期是主動脈弓再狹窄和RPA狹窄,重建的RPA在修復后經常出現狹窄,可能是由于重建困難,以及鄰近主動脈壓迫所致。根據Bi的文獻薈萃分析,約3/4的病例會在隨訪期間出現術后并發癥,RPA狹窄最常見(17/29,58.6%),其次是主動脈弓狹窄(13/29,44.8%),7例患兒接受了RPA狹窄的再干預治療,包括5例行外科手術修復和3例行球囊血管擴張術(其中1例患者同時接受了RPA的球囊血管擴張術和外科手術修復)[3]。所幸的是,本中心的4例患兒全部存活至今,無主動脈弓的再狹窄,僅病例2出現了嚴重的RPA狹窄,并接受了2次外科補片血管成形手術,故選擇主動脈內擋板技術修復時需謹慎注意APW到RPA的距離。Berry綜合征的一期手術修復是一個高風險和復雜的手術,但在有經驗的心臟中心是安全的。
Berry綜合征是一組非常罕見且復雜的心血管畸形,但可經心動超聲和CTA明確診斷。基于麻醉、體外循環和重癥監護的日益發展,可以在有經驗的心臟中心通過一期手術修復,本中心4例患兒術后無死亡,全部存活隨訪至今。本中心行右肺動脈前移重建后的2例患者的隨訪數據良好,右肺動脈發育良好,壓力梯度小。基于全球病例數少,應長期隨訪Berry綜合征患者,以期于優化這種罕見疾病的長遠療效。
利益沖突:無。
作者貢獻:施旭聰負責論文設計、撰寫及修改等;翁建彬、俞勁、馬曉輝參與數據整理與分析;石卓、俞建根、范祥明與論文審閱與修改。
Berry綜合征是一種極其罕見的復雜心血管畸形組合,1982年Berry首次進行了描述,其包括主-肺動脈窗(aortopulmonary window,APW)、右肺動脈異常起源于升主動脈(aortic origin of the right pulmonary artery,AORPA)、主動脈弓病變[包括主動脈弓中斷(interrupted aortic arch,IAA)、主動脈弓發育不良(hypoplastic aortic arch,HAA)、主動脈縮窄(coarctation of the aorta,CoA)],以及完整的室間隔[1]。Berry綜合征在先天性心臟病中的發生率約為0.046%,國內外文獻[2-3]有記載的病例不超過百例,通常表現出一種或多種臨床癥狀,如發紺、呼吸窘迫、心臟雜音、充血性心力衰竭等。大多數患者在出生后不久就死亡了,少數幸存者會出現嚴重的肺動脈高壓,因此早期、及時、準確的術前診斷和治療非常重要,而術前詳細評估該畸形的解剖特征對于制定手術策略非常關鍵。
基于麻醉、體外循環和重癥監護的日益發展,大多數中心提倡在新生兒期進行一期矯治手術[4-6]。本文通過回顧性分析浙江大學醫學院附屬兒童醫院收治的Berry綜合征患兒的臨床資料和手術策略,總結其解剖特征以及近遠期的隨訪結果,比較不同手術方式的優缺點,以期于優化這種罕見疾病的長遠療效。
臨床資料 4例患兒均通過經胸超聲心動圖和計算機斷層掃描血管成像(computerized tomographic angiography,CTA)明確診斷并根據其解剖特征進行如下分型:(1)根據Celoria和Patton的分類,確定IAA形態學診斷[7]:A型中斷發生在主動脈弓峽部,位于左鎖骨下動脈與動脈導管之間;B型中斷發生在左頸總動脈和左鎖骨下動脈之間;C型中斷發生在無名動脈和左頸總動脈之間。(2)APW類型是基于Mori的原始分類[8]:Ⅰ型是半月瓣緊上方的近端缺損;Ⅱ型是升主動脈更向頭部的遠端缺損;Ⅲ型是近端和遠端缺損相結合的完全缺損。(3)根據AORPA與APW的位置關系Berry將其分為2個亞型[1]:a型是指騎跨的右肺動脈(right pulmonary artery,RPA),血流動力學與升主動脈相關,但仍保持與主肺動脈和左肺動脈的解剖連續性;b型是另一種完全分離的RPA,在血流動力學和解剖學上都與升主動脈相關。
所有患兒均采用深低溫低流量順行選擇性腦灌注下進行主動脈弓重建。通過胸骨正中切口,插入2根動脈插管和上下腔插管建立體外循環:1根插入遠端升主動脈的右側,另1根插入動脈導管連接到降主動脈。轉流啟動后,左右肺動脈需立即阻斷,以防止肺循環充血。冷卻至合適的溫度后,阻斷升主動脈,將冷血心臟停搏液注入主動脈根部。然后拔除動脈導管插管,將升主動脈插管調整到無名動脈中,啟動深低溫低流量選擇性腦灌注。切斷縫扎動脈導管,充分游離升主動脈的分支及降主動脈,降主動脈上提后與主動脈弓部作端-側吻合,后壁直接吻合,前壁予以牛心包補片擴大成形。弓部修復完成后,將主動脈插管恢復到正常的弓位。然后通過主動脈前壁縱切口打開APW,進行徹底的檢查,分析APW的大小和位置、冠狀動脈的起源以及RPA的開口位置。
本中心采用了兩種手術方式來修復APW和AORPA:(1)主動脈內擋板技術:取合適大小的補片修補主動脈的缺損,并同時將RPA分隔至肺總動脈。(2)右肺動脈離斷再植技術:補片三明治法縫合修補APW,在主動脈后壁離斷RPA后用牛心包修補缺口,再將RPA移植到肺總動脈。本中心有3例患兒均使用了該手術方法,其中病例1的RPA在解剖原位移植到肺總動脈,病例3和4的RPA前移到主動脈的前方,肺總動脈的前壁取一矩形袢翻轉后與RPA后壁吻合,前壁用自體心包擴大成形。病例4術中的操作步驟見下圖1。
圖1
病例4 RPA前移重建的手術示意
a:A型IAA,升主動脈發出三個分支后中斷;b:RPA從主動脈的后壁發出;c:采用補片三明治法縫合修補APW;d:肺總動脈的前壁取一矩形袢翻轉后與RPA后壁進行吻合;e:用自體心包擴大成形RPA的前壁;f:重建后趴在主動脈前方的RPA;IAA:主動脈弓中斷;APW:主-肺動脈窗;AORPA:右肺動脈起源于升主動脈;RPA:右肺動脈
病例1,女,單胎40 W出生,因紫紺、咳嗽入院,診斷為:A型IAA,II型APW,AORPA與APW的位置形態為a型(圖2a)。手術年齡為28 d,手術體重為3.65 kg,主動脈阻斷時間為94 min,選擇性腦灌時間為50 min,術后ICU滯留時間10 d,術后呼吸機治療3 d。手術采用了RPA離斷再植技術(原位移植到肺總動脈)。術后隨訪至今已6年余,最后一次心超RPA壓力梯度為25 mm Hg(1 mm Hg=0.133 kPa),降主動脈流速為1.0 m/s。
圖2
所有患兒的影像資料
a:病例1的超聲切面可見特征性“蝴蝶征”影像;b:病例2的CTA三維成像顯示B型IAA;c:病例2的CT矢狀面成像顯示巨大的III型APW;d是病例3超聲切面的特征性“蝴蝶征”影像;e和f:病例4的CT成像顯示A型IAA、IIb型APW和AORPA;g:病例2術后隨訪時行CTA確診RPA再狹窄;h和f:分別是病例3和4術后超聲隨訪RPA發育良好無狹窄
病例2,女,因氣促、插管狀態下入院,診斷為:B型IAA,III型APW,AORPA與APW的位置形態為b型(圖2b、c)。手術年齡為8 d,手術體重為3.86 kg,主動脈阻斷時間為101 min,選擇性腦灌時間為79 min,術后呼吸機治療4 d,ICU期間發生了消化道大出血的并發癥,術后ICU滯留時間21 d,共住院50 d。手術采用了主動脈內擋板技術,肺總動脈前壁取一矩形袢并下翻用作主動脈內擋板的補片,然后用自體心包補片修補肺總動脈。然而患兒在隨訪期間發現RPA再狹窄(圖2g),術后定期常規超聲篩查提示RPA血流速度:2.7 m/s,壓差30 mm Hg,內徑0.3 cm,然后經CT血管造影(CTA)證實后,分別在初次手術后的第12個月和第22個月患兒接受了外科補片血管成形術。近期隨訪該患兒RPA和LPA都發育良好,最后一次心超RPA壓力梯度為9 mm Hg,降主動脈流速為1.6m/s。
病例3,女,單胎38 W+3出生,因黃疸入院,診斷為:A型IAA,II型APW,AORPA與APW的位置形態為b型(圖2d)。手術年齡為8 d,手術體重為3.05 kg,主動脈阻斷時間為108 min,選擇性腦灌時間為40 min,術后ICU滯留時間9 d,術后呼吸機治療2 d。手術采用了RPA離斷后前移重建技術。術后隨訪至今已2年余,最后一次心超RPA壓力梯度為3 mm Hg,降主動脈流速為1.3 m/s(圖2h)。
病例4,女,雙胎妊娠34 W+5早產兒,診斷為:A型IAA,II型APW,AORPA與APW的位置形態為b型(圖2e、f)。出生時體重僅2.30 kg,經過前列腺素E2開放動脈導管治療后,患兒生命體征平穩,遂繼續營養支持治療,待體重上升后再予以手術矯治,故其手術年齡為71 d,矯正胎齡為34 d,手術時體重為3.05 kg,主動脈阻斷時間為129 min,選擇性腦灌時間為34 min,術后呼吸機治療3 d,但因術后左側膈膨升經行膈肌折疊治療,術后ICU滯留時間為21 d,住院時間54 d。手術同樣采用了RPA離斷后前移重建技術。術后隨訪至今已半年余,最后一次心超RPA壓力梯度為4 mm Hg,降主動脈流速為1.5 m/s(圖2f)。
4例患者術后均順利出院并隨訪至今。病例3和病例4采用右肺動脈前移重建的方法,術后隨訪可見RPA發育佳,壓力梯度明顯較小。所有患兒均未出現喂養困難、聲帶麻痹、主動脈瓣上狹窄或主動脈弓再狹窄等其他并發癥。
本研究經浙江大學附屬兒童醫院倫理委員會批準(倫理批號:2023-IRB-0342-P-01),所有程序均符合機構和(或)國家研究委員會的倫理標準以及《赫爾辛基宣言》。
討論 Berry綜合征是一組極其罕見的先天性心血管畸形,其發病機制尚不明確,Berry等提出胚胎假說認為是APW影響了AORPA的形成,使流經主動脈弓的血流減少,從而導致HAA/CoA/IAA的發生[1]。由于其復雜性及危重性,如何在新生兒期給予正確的診斷和一期手術矯治仍然是臨床工作的難點。大多數患兒出生后早期即可出現嚴重的肺動脈高壓,APW和AORPA導致持續左向右分流,出現右肺動脈擴張,嚴重的甚至會壓迫氣道,而下半身的灌注則完全依賴于動脈導管[2]。多數學者建議在新生兒期對這些患者進行手術矯正,以保護肺血管床避免形成不可逆的肺高血,并盡早恢復下半身的正常血供[5-6,9]。根據Bi等[3]的Meta分析,大多數Berry綜合征為男性(49/73,67.1%),絕大部分可在產前或出生后3個月內明確診斷(56/81,69.1%),合并A型IAA(67/76,88.2%)、II型APW(56/71,78.9%)和b型AORPA為主(73/86,84.9%),未手術患者的死亡率為100%,死亡年齡的中位數是1個月。本中心的4例患兒除性別均是女性之外,其他臨床特征都與文獻描述相符,且均在新生兒期得到明確診斷,及全部完成一期手術矯治,術后存活率100%。
經胸超聲心動圖是出生后評估Berry綜合征的初始工具,這是一種安全且侵入性較小的方式,但由于有限的成像平面和視野,可能會出現AORPA的漏診,聯合CTA,出現RPA起源于升主動脈、左肺動脈起源于主肺動脈、APW位于主動脈和肺動脈之間,呈現“蝴蝶征”這種特征性影像時需高度警惕Berry綜合征的診斷[10]。本中心的4例患兒通過超聲聯合CTA,在新生兒期得到了明確診斷,并及時予以合適的治療策略。
手術修復的策略有兩種:(1)分期修復:第一階段放置肺動脈環縮帶控制肺血流,患兒通氣狀況改善后再接受完整的手術修復,Ghelani等[11-12]建議早產兒或小于胎齡兒應考慮分期修復。但隨著體外監護的日益發展,分期手術可酌情選擇。本中心病例4是早產兒且低體重,但經內科治療后患兒生命體征平穩,遂繼續營養支持治療,故其手術年齡為71 d,術后隨訪肺動脈壓力逐漸下降正常。(2)一期手術矯治:通過關閉APW,將RPA連接到肺總動脈,并建立主動脈弓連續性來實現的,大多數文獻報道一期矯治手術可獲得令人滿意的結果,且兩組之間術后1個月內的死亡率或1年隨訪期內的存活時間沒有顯著性差異[3,5,13]。矯治手術的解剖特點主要集中在APW的大小和位置,RPA異常起源的位置,以及IAA的類型和范圍,重建無張力且連續性的RPA和主動脈弓是影響手術結果的重要因素。本中心4例患兒均采用主動脈后壁端側吻合、前壁人工材料擴大成形修復主動脈弓,促進潛在的血管生長,術后均無主動脈弓再狹窄,無持續性肺動脈高壓,總手術風險可能類似于單純性IAA,近期和中期血流動力學和臨床結果均令人滿意。
本中心采用了2種手術方法重建RPA的連續性:(1)主動脈內擋板技術,技術要點是精確修整主動脈內補片的大小,防止左心室流出道和RPA的梗阻。胡等提出所有Ⅱa型APW和AORPA的Berry綜合征患兒均可采用該方法,但僅適用于部分Ⅱb型,這取決于APW到RPA的距離[4]。然而本中心的病例2(B型IAA和Ⅲ型APW)采用了該手術方法,術后出現了嚴重的RPA起點處的狹窄,說明該技術可能不適合Ⅲ型APW,考慮其補片較大,升主動脈血壓高,補片會被推向肺動脈導致壓迫狹窄。(2)RPA離斷再植技術,是孤立性RPA起源異常患兒最常采用的手術方法,可用于任何類型的Berry綜合征,無論RPA的位置或APW的大小。本中心其余3例均使用了該手術方法,其中病例1的RPA解剖原位移植到肺總動脈,病例3和4的RPA前移到主動脈的前方,與LeCompte操作相似,肺總動脈的前壁取一矩形袢翻轉后與RPA后壁吻合,前壁用自體心包擴大成形。與主動脈內擋板技術相比較,該方法破壞了RPA和MPA之間的原有連續性,且手術操作更復雜[14-16]。RPA前移重建的手術方法在個別病例報道中也獲得了良好的術后效果,Chang等[17]對1例伴有APW、AORPA、IAA和室間隔缺損的足月女嬰采用了RPA前移重建的一期手術矯治,術后氣道壓迫立即完全緩解,術后隨訪35個月CTA證實RPA發育良好。本中心2例采用RPA前移重建的患兒比1例采用RPA原位重建的患兒,獲得了更好的RPA發育,壓力梯度更小。
術后并發癥早期是院內死亡,遠期是主動脈弓再狹窄和RPA狹窄,重建的RPA在修復后經常出現狹窄,可能是由于重建困難,以及鄰近主動脈壓迫所致。根據Bi的文獻薈萃分析,約3/4的病例會在隨訪期間出現術后并發癥,RPA狹窄最常見(17/29,58.6%),其次是主動脈弓狹窄(13/29,44.8%),7例患兒接受了RPA狹窄的再干預治療,包括5例行外科手術修復和3例行球囊血管擴張術(其中1例患者同時接受了RPA的球囊血管擴張術和外科手術修復)[3]。所幸的是,本中心的4例患兒全部存活至今,無主動脈弓的再狹窄,僅病例2出現了嚴重的RPA狹窄,并接受了2次外科補片血管成形手術,故選擇主動脈內擋板技術修復時需謹慎注意APW到RPA的距離。Berry綜合征的一期手術修復是一個高風險和復雜的手術,但在有經驗的心臟中心是安全的。
Berry綜合征是一組非常罕見且復雜的心血管畸形,但可經心動超聲和CTA明確診斷。基于麻醉、體外循環和重癥監護的日益發展,可以在有經驗的心臟中心通過一期手術修復,本中心4例患兒術后無死亡,全部存活隨訪至今。本中心行右肺動脈前移重建后的2例患者的隨訪數據良好,右肺動脈發育良好,壓力梯度小。基于全球病例數少,應長期隨訪Berry綜合征患者,以期于優化這種罕見疾病的長遠療效。
利益沖突:無。
作者貢獻:施旭聰負責論文設計、撰寫及修改等;翁建彬、俞勁、馬曉輝參與數據整理與分析;石卓、俞建根、范祥明與論文審閱與修改。