

肺癌是全球癌癥相關死亡的主要原因[1]。非小細胞肺癌(non-small cell lung cancer,NSCLC)約占所有肺癌的85%,其主要組織學亞型是肺腺癌(lung adenocarcinoma,LUAD),約占肺癌發病率的40%[2]。盡管LUAD的靶向治療和免疫治療取得了重大進展,但由于疾病的隱匿性和缺乏特異性,大多數患者在確診時已處于晚期,5年總生存率仍低于20%[3]。因此,迫切需要探索新的生物標志物和可靠的預后預測模型來改善LUAD患者的生存結局。
晝夜節律系統協調人體生理和行為節律以適應24 h生理周期[4]。晝夜節律基因主要分為兩類:核心時鐘基因和時鐘控制基因,前者控制人體生理晝夜節律,后者調節核心時鐘基因的表達[5]。許多研究[4,6-7]表明,晝夜節律紊亂不僅會導致代謝、心血管和免疫功能障礙,而且還與患癌風險和較差的預后有關。隨著現代醫學的發展,在高通量RNA測序技術和人工智能的輔助下,生物信息學結合機器學習算法已被廣泛應用于基因組和蛋白質組研究,越來越多疾病的篩查、診斷和預后模型被開發出來[8]。單細胞RNA測序技術(single cell sequencing,scRNA-seq)被用于分析腫瘤微環境(tumor microenvironment,TME)的細胞類型和腫瘤異質性等相關生物學信息[9],能夠在單細胞水平上檢測基因表達量,從而有助于剖析腫瘤細胞群體中基因表達的異質性[10]。借助這一優勢,scRNA-seq和生物信息學數據分析方法的發展為揭示TME中不同細胞群體的分子特征提供了前所未有的機會[3]。
1 資料與方法
本研究首先對癌癥基因組圖譜(The Cancer Genome Atlas,TCGA)的LUAD晝夜節律基因表達量和臨床病理數據進行全面分析。然后通過Cox回歸和3種不同的機器學習算法篩選出與LUAD預后相關的特征晝夜節律基因,構建LUAD預后的晝夜節律基因預測模型,并驗證該模型在外部數據集的穩定性。此外,我們還評估了該模型與免疫細胞和免疫檢查點基因的關聯。最后在scRNA-seq分析的基礎上,探討預后相關的特征晝夜節律基因與TME中不同免疫細胞群體之間的分子特征。研究流程見圖1。
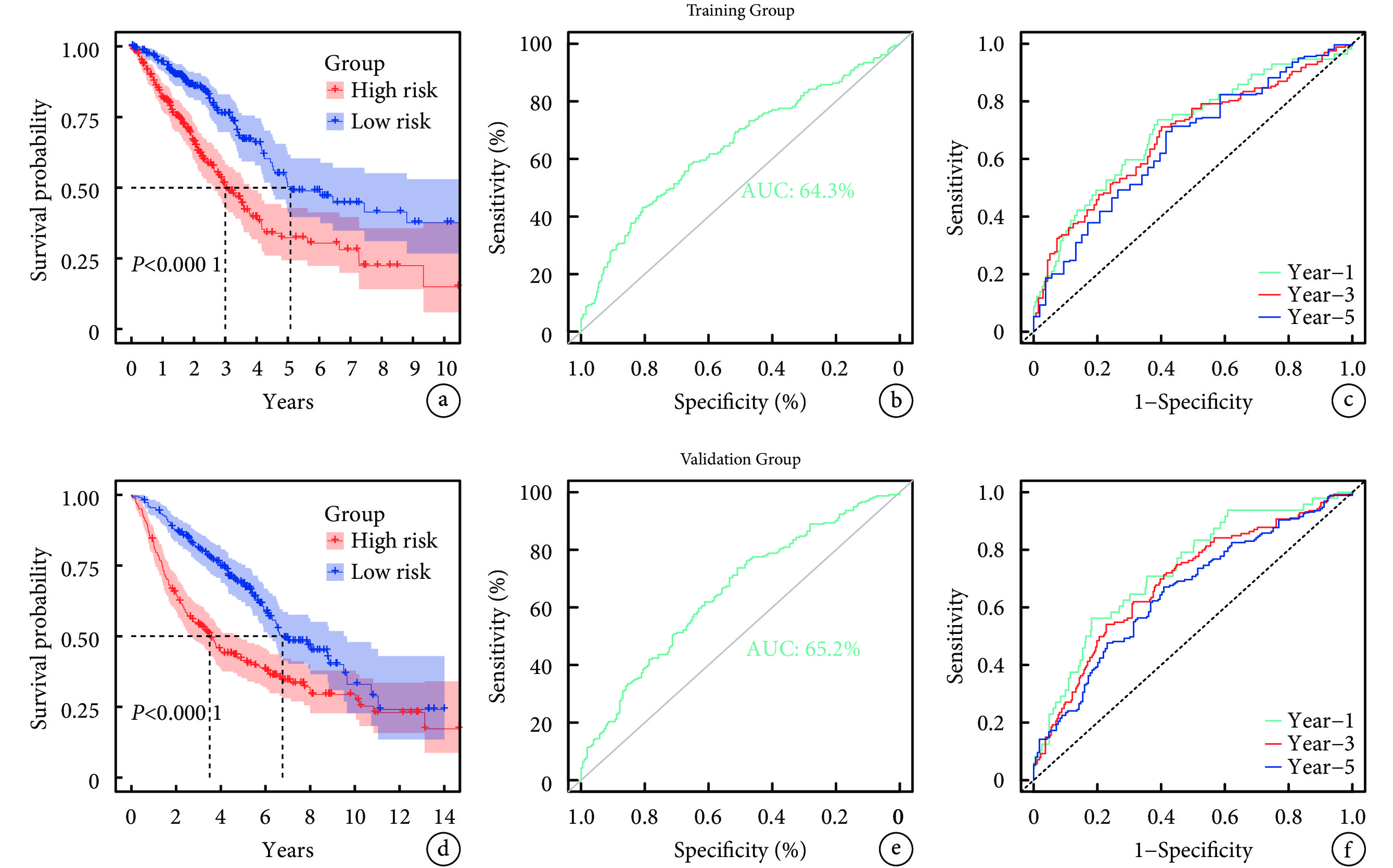 圖1
研究流程圖
圖1
研究流程圖
1.1 數據來源
晝夜節律基因集(210個基因)于2023年6月27日從晝夜節律基因數據庫(Circadian Genes Database,CGDB;http://cgdb.biocuckoo.org)得到,同期在TCGA數據庫(https://portal.gdc.cancer.gov)獲得LUAD RNA-Seq數據和臨床病理數據(524例癌癥樣本,58例癌旁正常組織樣本),篩選出LUAD患者晝夜節律基因表達量,并經過log2的轉化。從Gene Expression Omnibus(GEO)數據庫(https://www.ncbi.nlm.nih.gov/geo)獲得外部驗證數據集GSE68465(443例癌癥樣本,19例癌旁正常組織樣本)。
1.2 晝夜節律差異基因篩選、突變及拷貝數變異分析
從TCGA數據庫下載LUAD基因突變數據和拷貝數變異(copy number variations,CNVs)數據,與晝夜節律基因相對應,分別通過R-4.3.0軟件“maftools”和“Rcircos”包處理并可視化。采用“DESeq2”包以P≤0.05和|log2(fold change)|>1的標準篩選晝夜節律差異表達基因(differentially expressed genes,DEGs),并使用“pheatmap”“ggplot2”和“psych”包繪制火山圖和基因與臨床性狀相關性熱圖。
1.3 功能富集分析和構建蛋白質相互作用網絡
使用“enrichplot”“org.Hs.eg.db”和“clusterProfiler”包對DEGs進行基因本體功能富集分析(gene ontology,GO)、京都基因與基因組百科全書通路分析(Kyoto Encyclopedia of Genes and Genomes,KEGG)和基因集富集分析(gene set enrichment analysis,GSEA),使用“ggplot2”包對結果進行可視化。通過The STRING網站(www.string-db.org)以最低相關分數0.400為閾值生成DEGs蛋白質相互作用網絡(protein-protein interaction network,PPI)[11]。數據以TSV格式從數據庫下載,并用Cytoscape_v3.9.0軟件可視化。P≤0.05為差異有統計學意義。
1.4 構建風險評分預測模型
將訓練組524例LUAD樣本的晝夜節律DEGs表達量和生存信息合并,剔除15例無總生存期(overall survival,OS)及生存狀態的患者,余509例LUAD樣本依次納入Cox回歸和3種機器學習算法[最小絕對收縮和選擇算子(LASSO)回歸、支持向量機遞歸特征消除(SVM-RFE)、隨機森林]。選擇4種算法計算DEGs的交集基因,最終構建風險評分(RiskScore)預測模型。RiskScore預測模型計算公式:RiskScore=Exp1×C1+Exp2×C2+…+Expn×Cn(Exp為預后特征基因的表達量,C為LASSO回歸分析得到的回歸系數,n為交集基因的數目)。
根據上面的公式計算每例樣本的RiskScore,以RiskScore中位數為截斷值將LUAD樣本分為低風險組和高風險組,P≤0.05為差異有統計學意義。
1.5 評價風險預測模型和繪制列線圖
首先繪制Kaplan-Meier生存曲線比較低風險組和高風險組的OS。其次通過“timeROC”包繪制時間依賴性受試者工作特征(receiver operating characteristic,ROC)曲線,計算出訓練組中模型預測1、3、5年OS率的曲線下面積(area under the curve,AUC),評價預測風險模型的準確性。最后將兩組的臨床病理因素和RiskScore納入Cox回歸分析,并構建列線圖。利用校準曲線和ROC曲線評價生存預測的準確性。使用GSE68465數據集驗證構建的預測模型。
1.6 評估腫瘤微環境的免疫學特征
通過應用“CIBERSORT”包對22種不同免疫細胞的浸潤狀態進行評估,以探討RiskScore與免疫細胞浸潤之間的關系。此外,從相關文獻中檢索出46個常見的免疫檢查點,探索RiskScore與46個免疫檢查點基因之間的聯系[12]。P≤0.05為差異有統計學意義。
1.7 藥物敏感性分析
相關基因表達和藥物數據從CellMiner數據庫(https://discover.nci.nih.gov/cellminer/home.do)下載。使用臨床實驗室驗證以及國家藥品監督管理局標準認證相結合的方法對藥物數據進行篩選。然后,將預后特征晝夜節律基因的表達數據與藥物數據合并,進行Pearson相關性檢驗,確定其相關性和藥物敏感性。
1.8 特征晝夜節律基因的單細胞群體分子特征
為進一步驗證特征晝夜節律基因與TME的關系,采用來自GSE149655數據集(GSM4506699、GSM4506701)的2例LUAD 10×scRNA-seq數據,并利用“Seurat”“patchwork”和“dplyr”包進行數據整理、標準化以及主成分分析(principal component analysis,PCA)。首先通過以下篩選標準保留高質量scRNA-seq數據:(1)剔除線粒體基因超過25%的細胞;(2)提取表達>200個且<
2 結果
2.1 晝夜節律基因的差異表達和遺傳變異模式
遺傳變異數據與晝夜節律基因一一對應后得到的基因突變和CNVs數據(201個基因,447例樣本)在LUAD中的體細胞突變頻率為68.01%(304/447);見圖2a。從TCGA數據庫收集的LUAD患者晝夜節律基因以P≤0.05和|log2(fold change)|>1為閾值,篩選出57個DEGs,包括37個上調基因和20個下調基因,晝夜節律DEGs在染色體的位置見圖2b。DEGs與臨床性狀相關性熱圖、DEGs的火山圖可視化結果見圖2c~d。
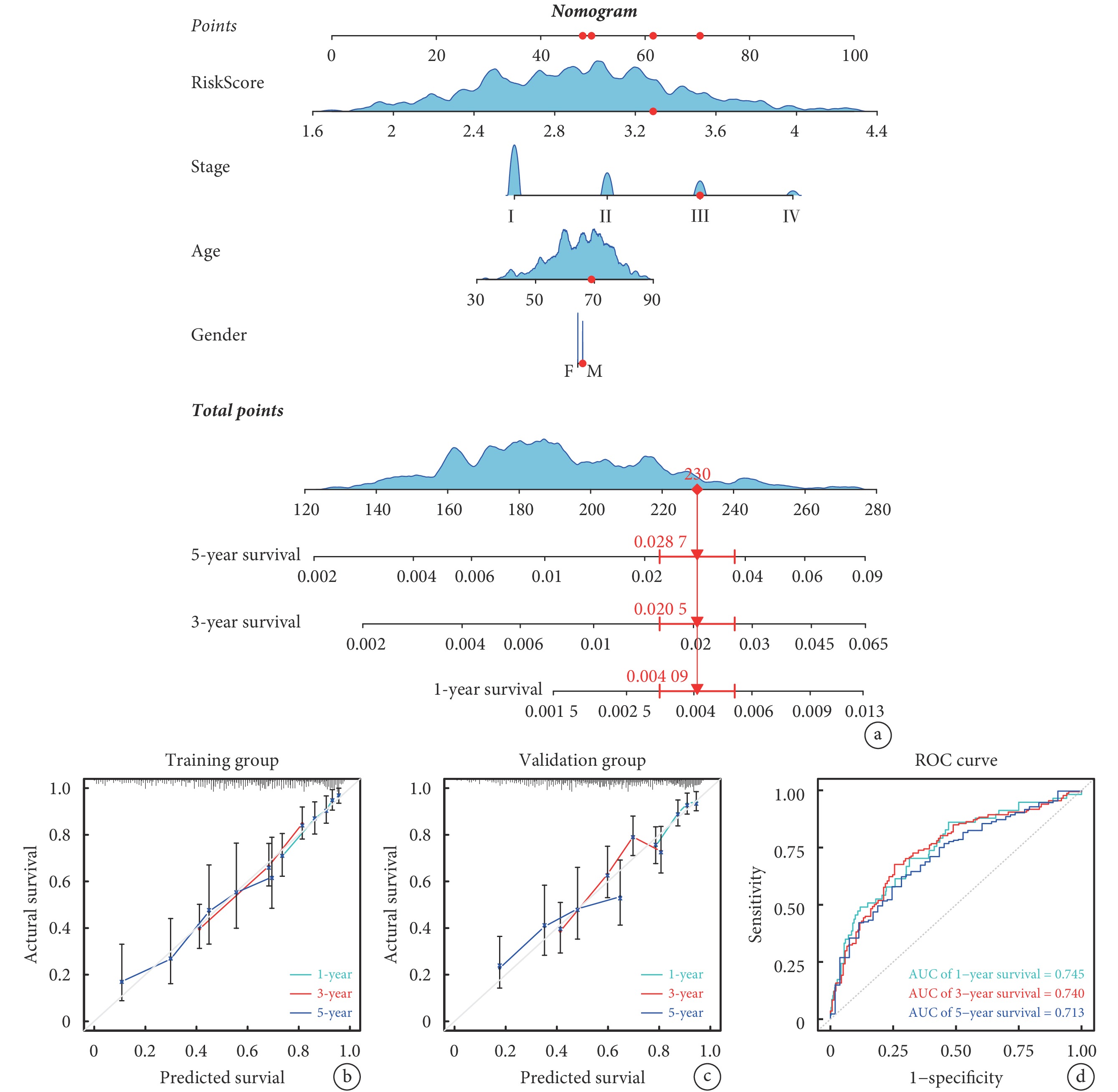 圖2
肺腺癌晝夜節律基因的分子特征和表達變異
圖2
肺腺癌晝夜節律基因的分子特征和表達變異
a:447 例肺腺癌患者晝夜節律基因的突變頻率,每列代表 1 例患者,頂部的條形圖代表腫瘤突變負荷,右側的數字代表每個基因的突變頻率,右側的條形圖顯示了每種基因的比例,下面堆疊的條形圖顯示了每個樣本中轉化的百分比;b:差異表達基因在23條染色體上的位置;c:差異表達基因與臨床特征的相關性熱圖;d:晝夜節律差異表達基因火山圖
2.2 功能富集分析和差異表達基因蛋白質相互作用網絡的構建
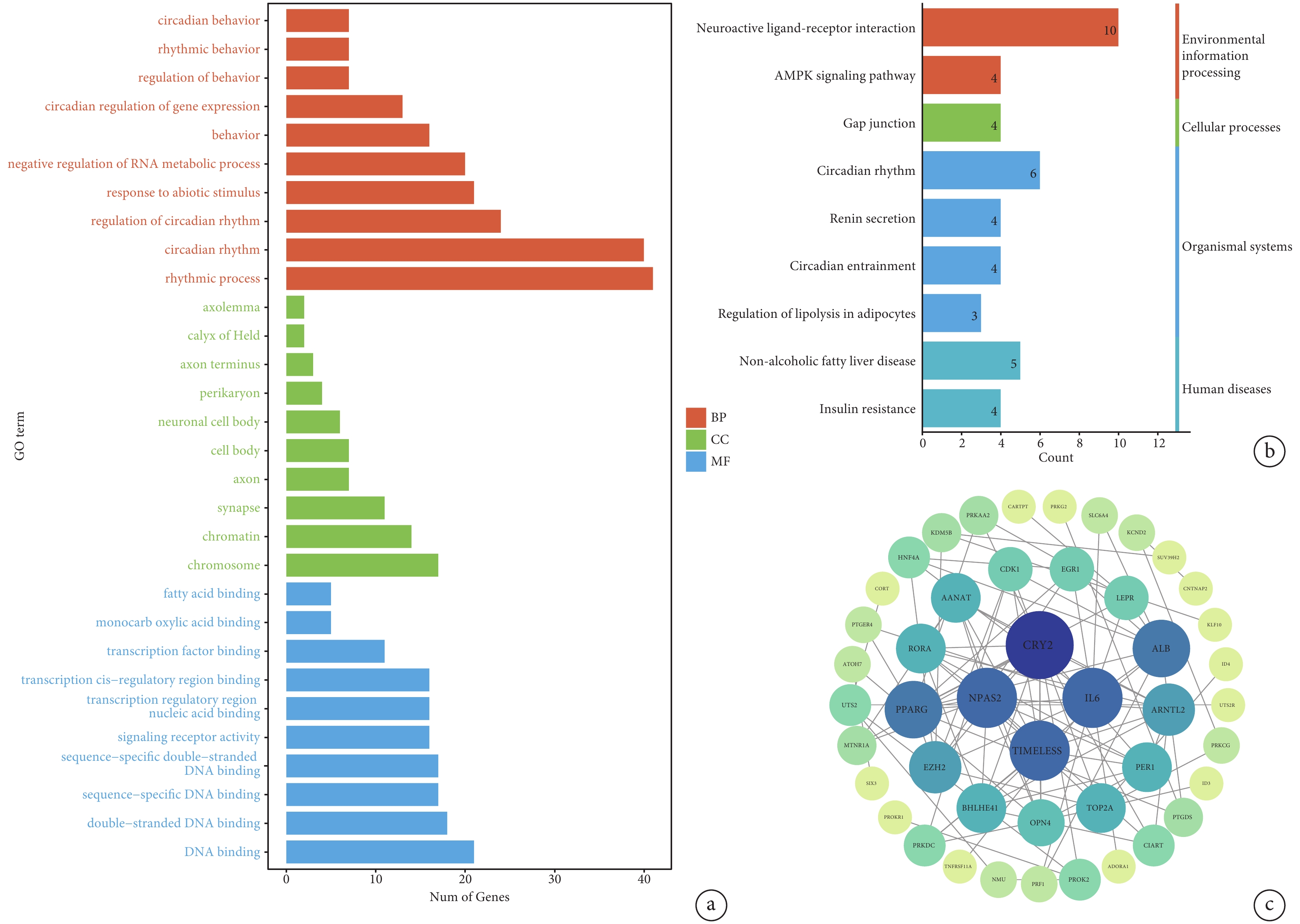
為探索LUAD中晝夜節律基因的相關生物學功能和途徑,對上述57個DEGs進行GO及KEGG分析,富集度最高的GO分類和KEGG通路見圖3a~b。GO分析顯示,DEGs主要富集在晝夜節律行為、RNA代謝的負調控、序列特異性DNA結合等相關生物學功能;KEGG分析顯示,DEGs主要富集在AMPK信號通路、脂肪細胞脂解的調控通路和神經活性配體-受體相互作用通路等。將P≤0.05的晝夜節律基因納入GSEA分析,其主要富集于cGMP-PKG信號通路、脂質與動脈粥樣硬化和JAK-STAT信號通路等相關生物學過程,進一步驗證了遺傳物質調控和脂肪酸等能量物質代謝與腫瘤進展的密切關系。
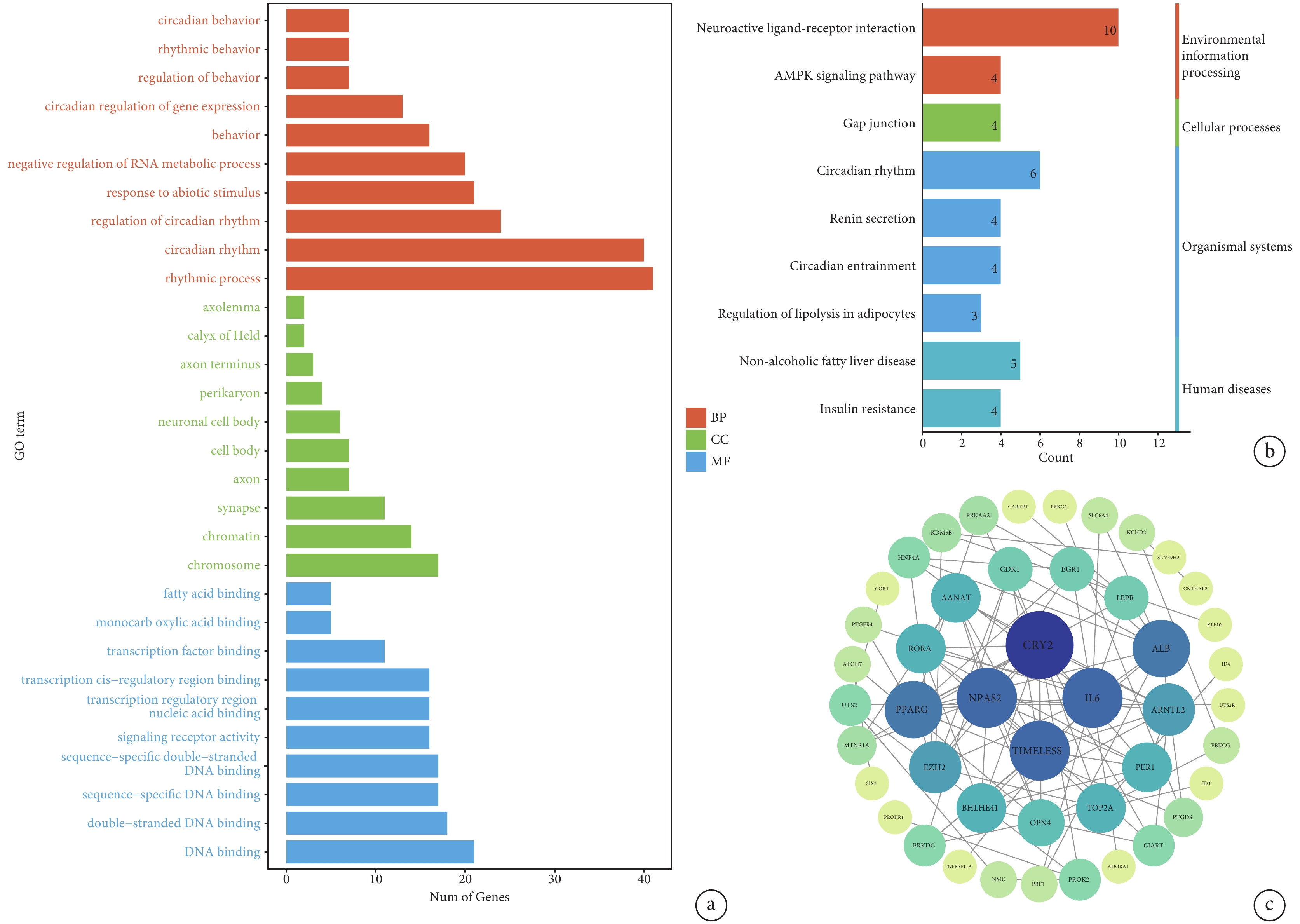 圖3
功能富集分析和 PPI 網絡的構建
圖3
功能富集分析和 PPI 網絡的構建
a:GO 分析,對前 10 個富集類別(生物過程、細胞成分和分子功能)進行可視化;b:KEGG 分析,顯示前 9 個富集通路;c:PPI 網絡構建,基于 PPI 網絡的前 46 個節點差異表達基因,使用 Degree 算法選擇基因,顏色越深表示基因關聯的節點越多;PPI:蛋白質相互作用網絡
在去除不與其他節點交互的DEGs后,通過Degree算法構建了46個節點的PPI網絡,展示晝夜節律DEGs蛋白質之間的相互作用;見圖3c[13]。以上分析結果解釋了基因層面的關系,也為將來尋找與預后相關生物學標志物的潛在靶點提供了研究思路。
2.3 篩選預后特征基因并構建風險評分預測模型
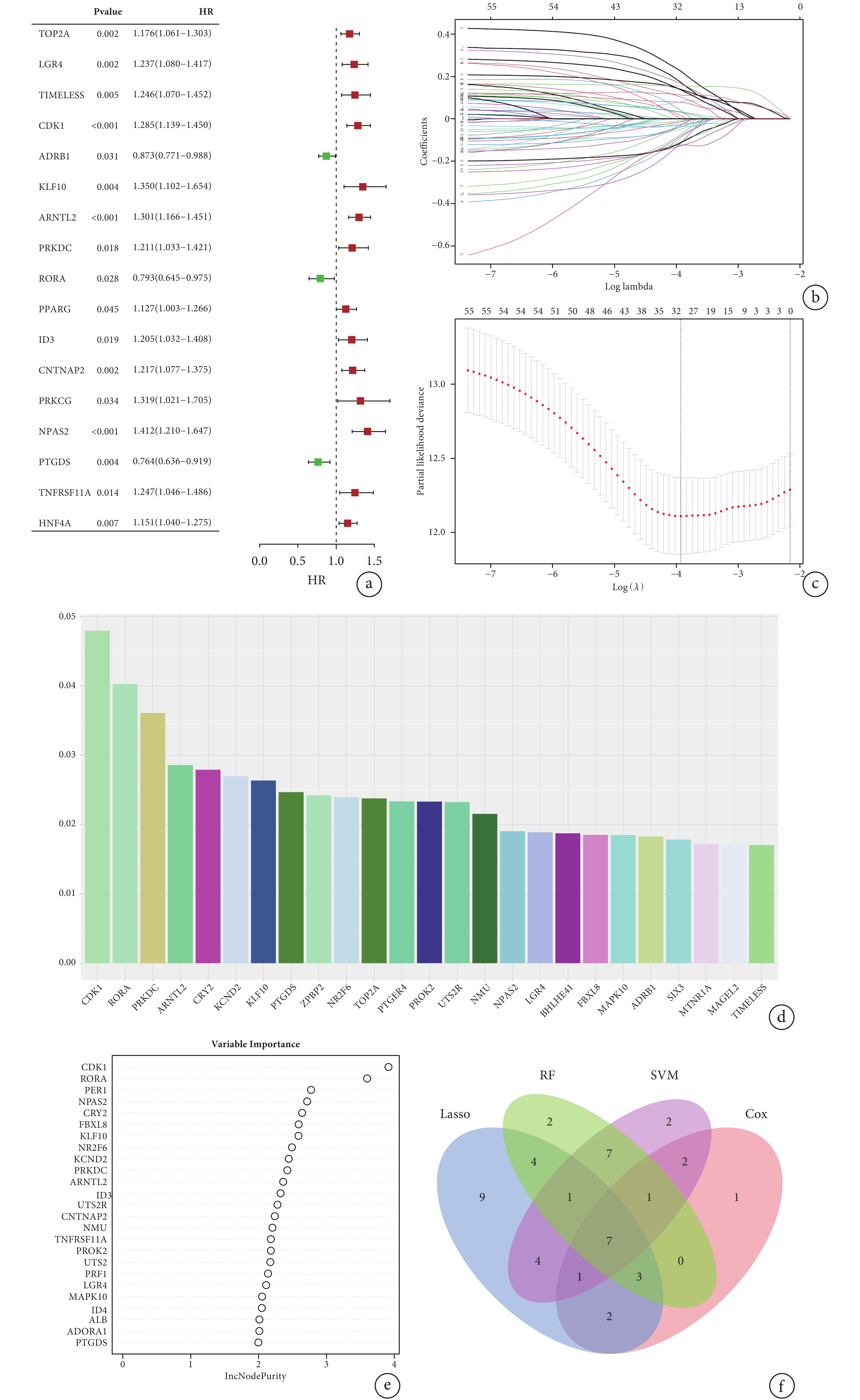
將上述57個DEGs分別納入Cox回歸、LASSO回歸、SVM-RFE和隨機森林。Cox回歸得到17個與預后獨立相關的基因(P<0.05);見圖4a。通過LASSO回歸的降維分析,獲得31個特征基因;見圖4b~c。應用SVM-RFE方法根據其重要性評分鑒定出25個準確率最高的基因;見圖4d。通過隨機森林算法對DEGs重要性評分排序后,識別出25個候選基因;見圖4e。最后,將每種方法篩選的DEGs取交集后的7個預后相關特征基因(LGR4、CDK1、KLF10、ARNTL2、RORA、NPAS2、PTGDS)可視化為Venn圖;見圖4f。根據7個基因的表達量和風險系數構建RiskScore模型:RiskScore=LGR4×
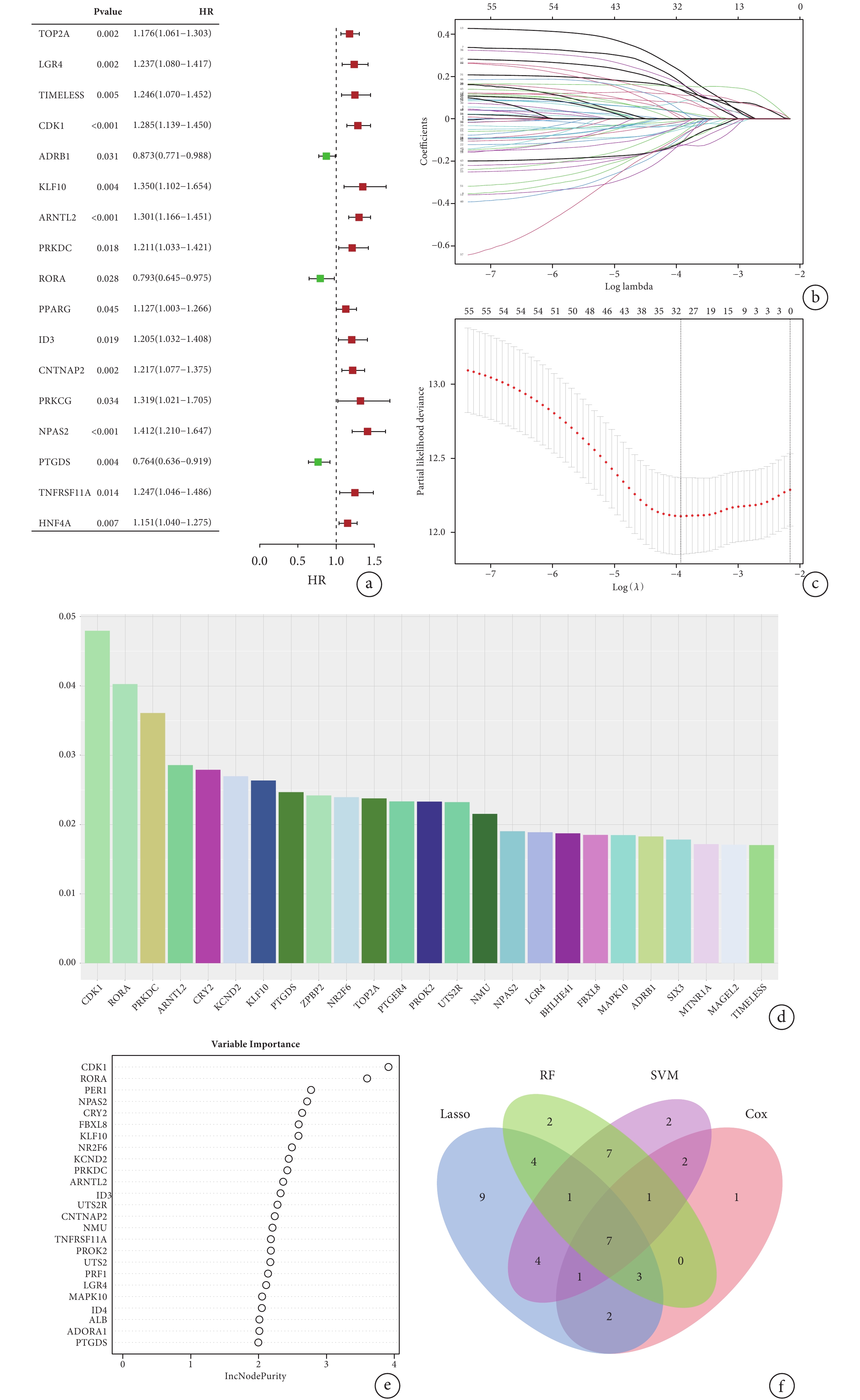 圖4
機器學習算法篩選生物標志物
圖4
機器學習算法篩選生物標志物
a:Cox 分析顯示 17 個基因與預后顯著相關;b~c:LASSO 回歸分析用于篩選晝夜節律基因的生物標志物,虛線標注處為Log(
2.4 風險評分與臨床病理特征的關系
以訓練組LUAD樣本的RiskScore中位數為截斷值將訓練組樣本分為低風險組(n=255)和高風險組(n=254)。不同風險組患者表現出不同的臨床病理特征。病理分期(P<0.001)、T分期(P=0.02)和N分期(P<0.001)均與RiskScore相關,然而年齡(P=0.59)和性別(P=0.31)與RiskScore無關。LGR4(R=0.419,P<0.001)、CDK1(R=0.710,P<0.001)、KLF10(R=0.499,P<0.001)、ARNTL2(R=0.736,P<0.001)和NPAS2(R=0.523,P<0.001)與RiskScore呈正相關,RORA(R=?0.204,P<0.001)和PTGDS(R=?0.356,P<0.001)與RiskScore呈負相關。
2.5 檢驗風險評分預測模型性能并繪制列線圖
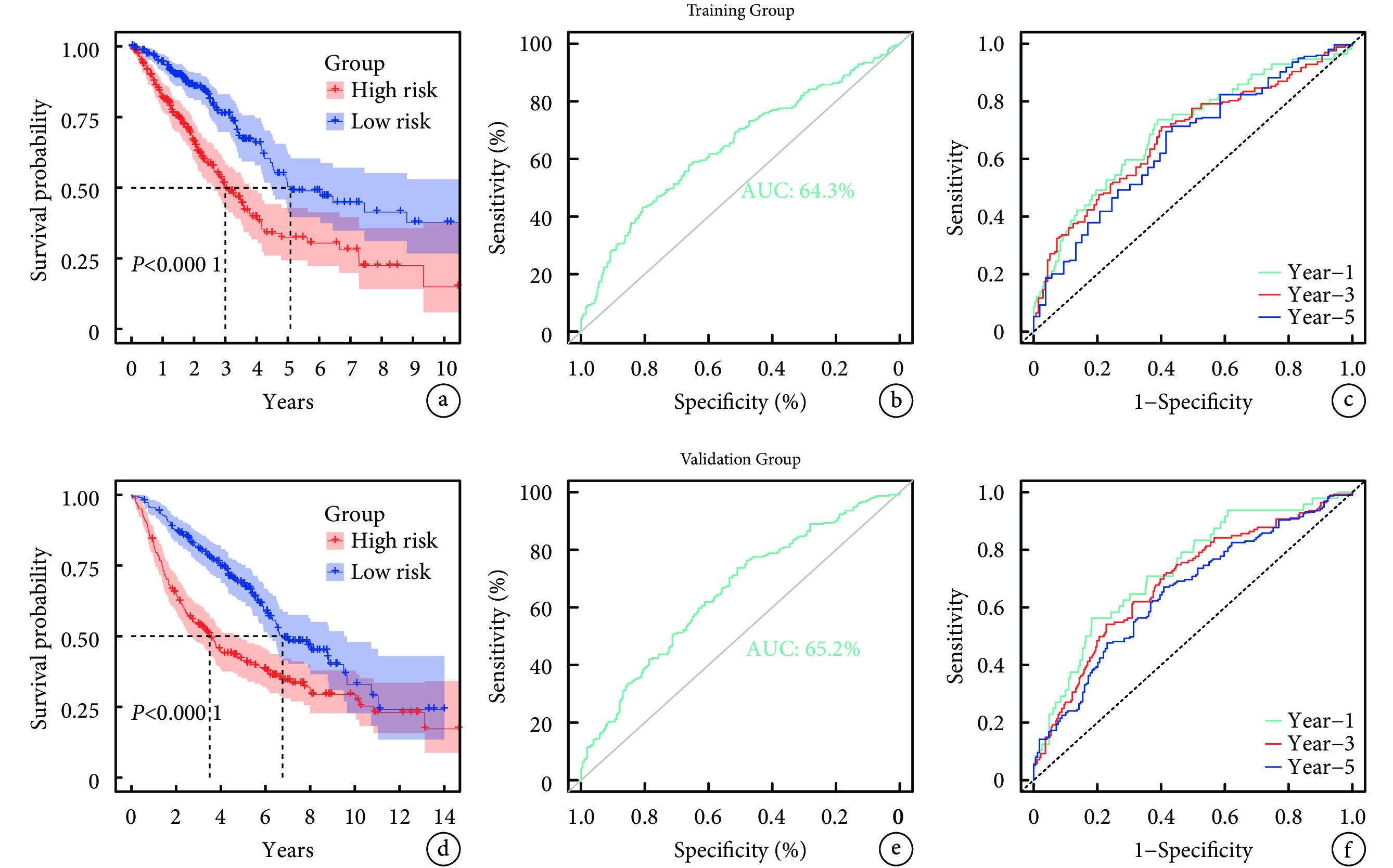
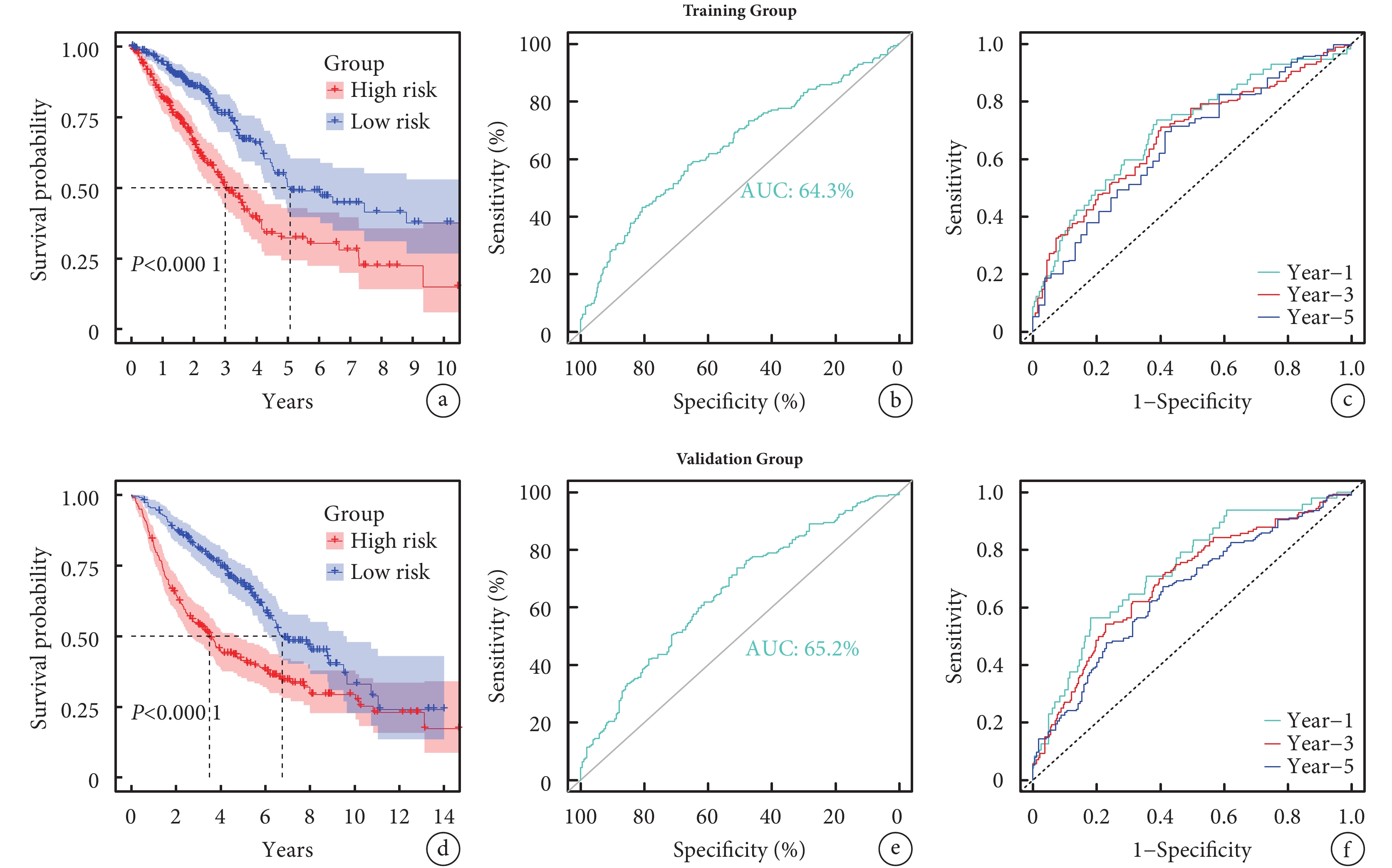
Kaplan-Meier生存曲線顯示,訓練組樣本中,高風險和低風險組OS差異有統計學意義(P<0.001),表明高風險組比低風險組預后差;見圖5a。為確保RiskScore預測模型的穩定性,繪制ROC曲線和時間依賴性ROC曲線,ROC曲線的AUC值為0.643,時間依賴性ROC曲線顯示,預測1、3、5年OS率的AUC值分別為0.702、0.680、0.654,表明模型具有良好的準確性;見圖5b~c。驗證組數據集剔除1例無OS的樣本后,將剩余442例樣本以相同方法分為低風險組(n=221)和高風險組(n=221),然后繪制Kaplan-Meier生存曲線、ROC曲線及時間依賴性ROC曲線,兩組OS差異仍有統計學意義(P<0.001)。ROC曲線的AUC值為0.643,時間依賴性ROC曲線顯示,預測1、3、5年OS率的AUC值分別為0.730、0.688、0.653;見圖5d~f。上述結果說明該模型在驗證組中仍具有較好的預測性能。
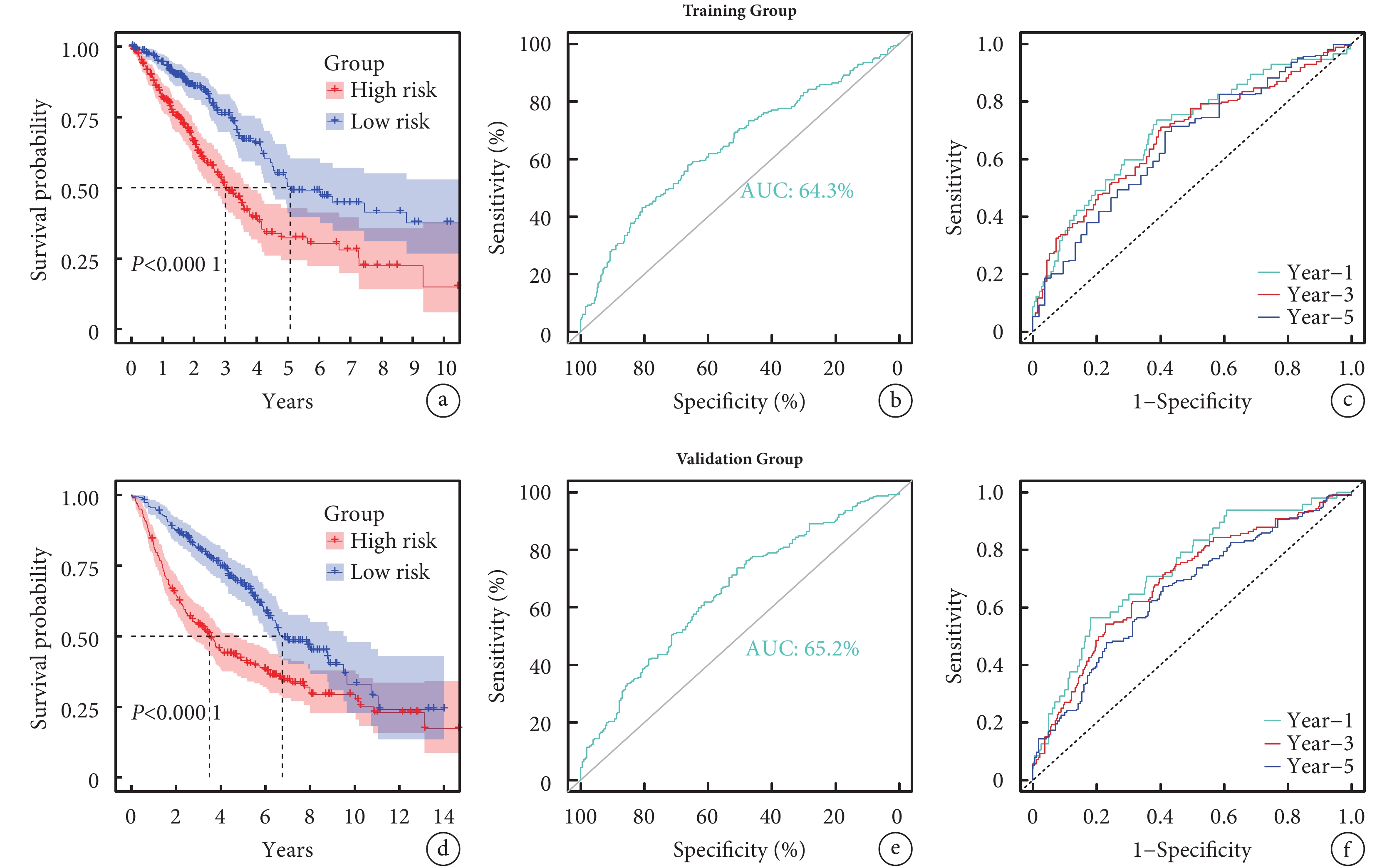 圖5
評估RiskScore模型的預測價值
圖5
評估RiskScore模型的預測價值
a~c:分別為訓練組的Kaplan-Meier生存曲線、ROC曲線和時間依賴性ROC曲線;d~f:分別為驗證組的Kaplan-Meier生存曲線、ROC曲線和時間依賴性ROC曲線;ROC:受試者工作特征;AUC:曲線下面積
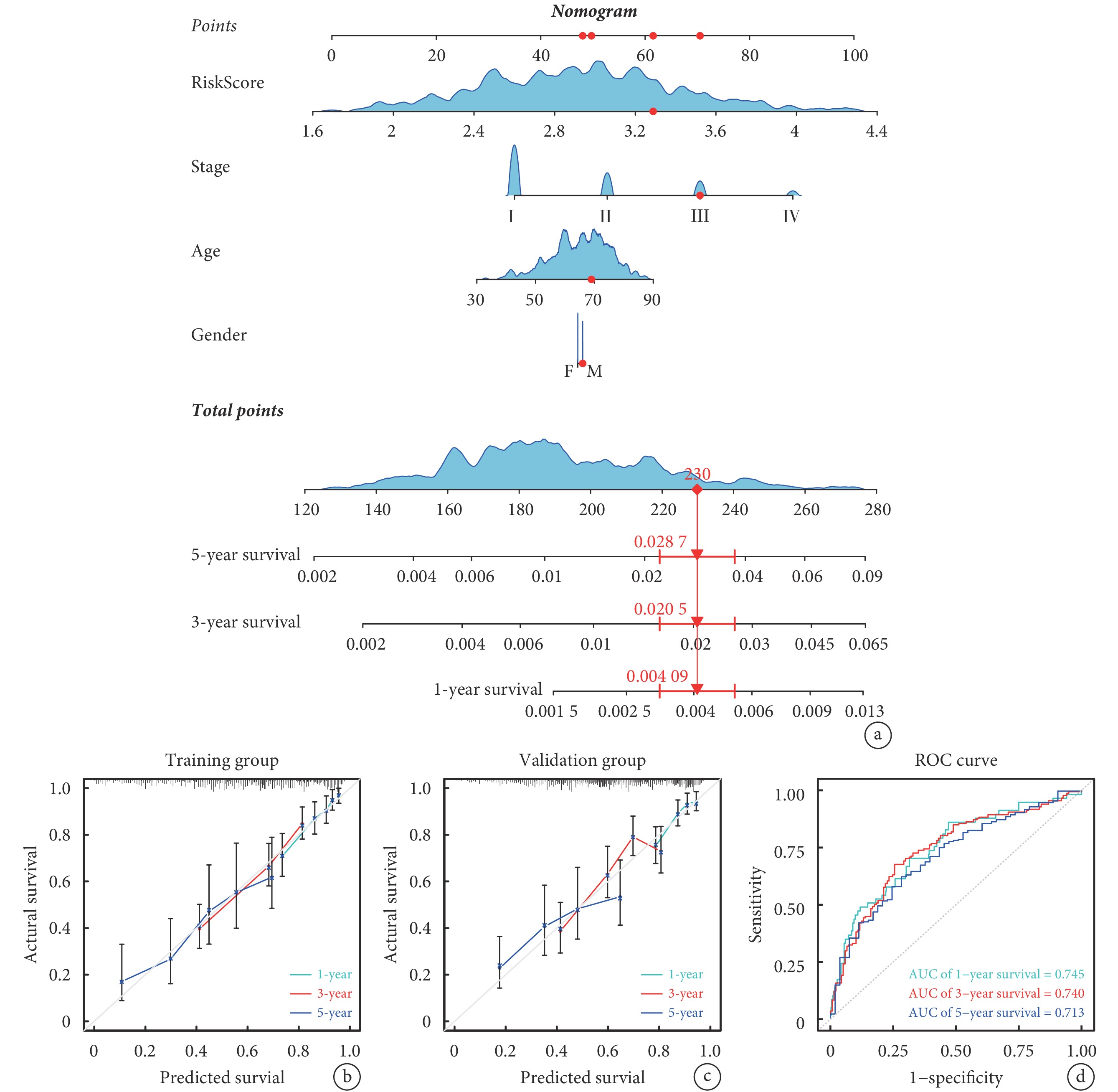
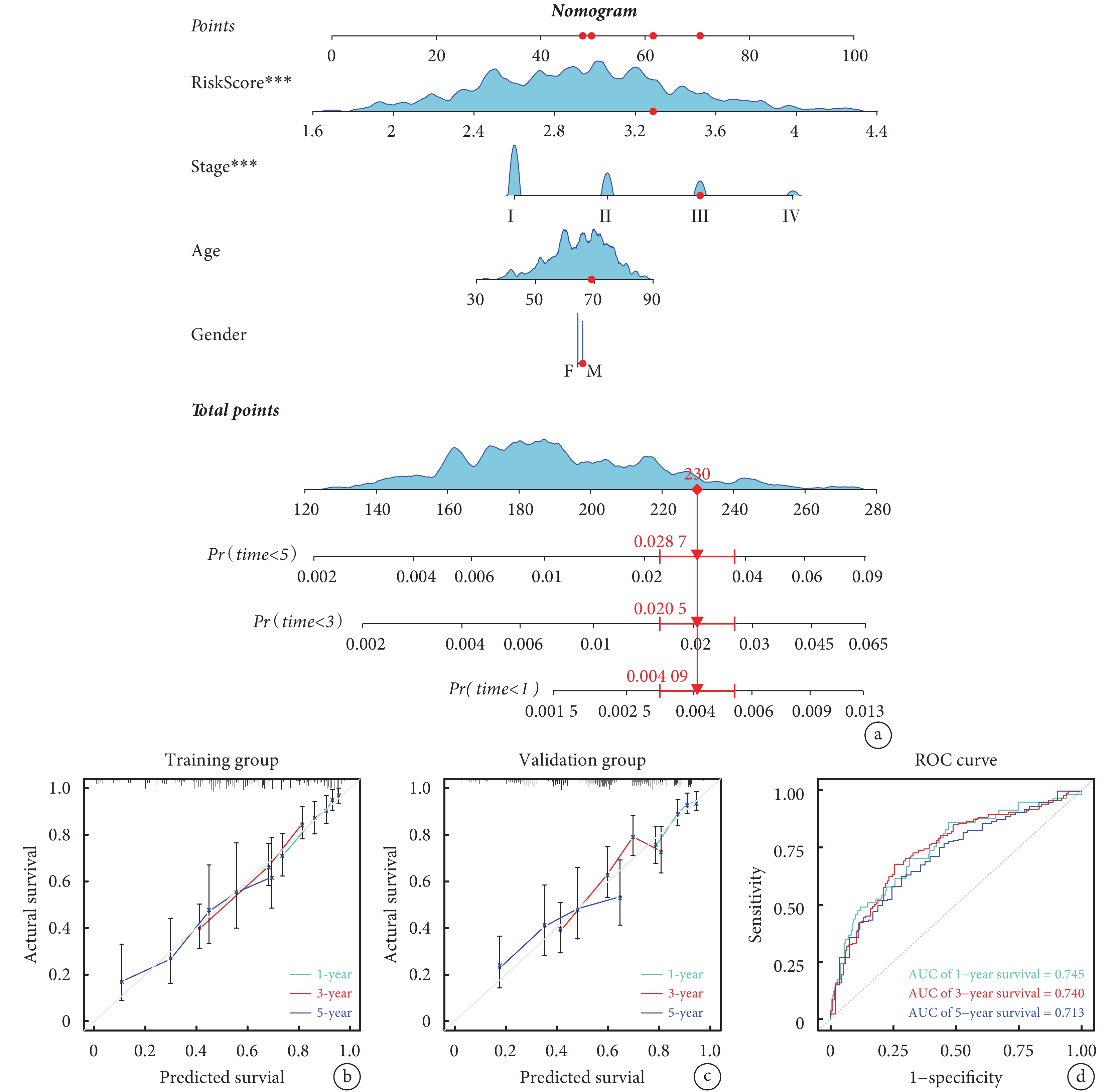
通過評估基于晝夜節律基因的風險模型在臨床實踐中的有效性,將RiskScore及臨床病理特征納入Cox回歸分析,篩選LUAD患者OS的獨立危險因素,并構建列線圖;見圖6a。該個體化預測模型可預測LUAD患者1、3、5年OS率,其AUC值分別為0.745、0.740和0.713。校準曲線和ROC曲線顯示,列線圖預測結果可靠;見圖6b~d。
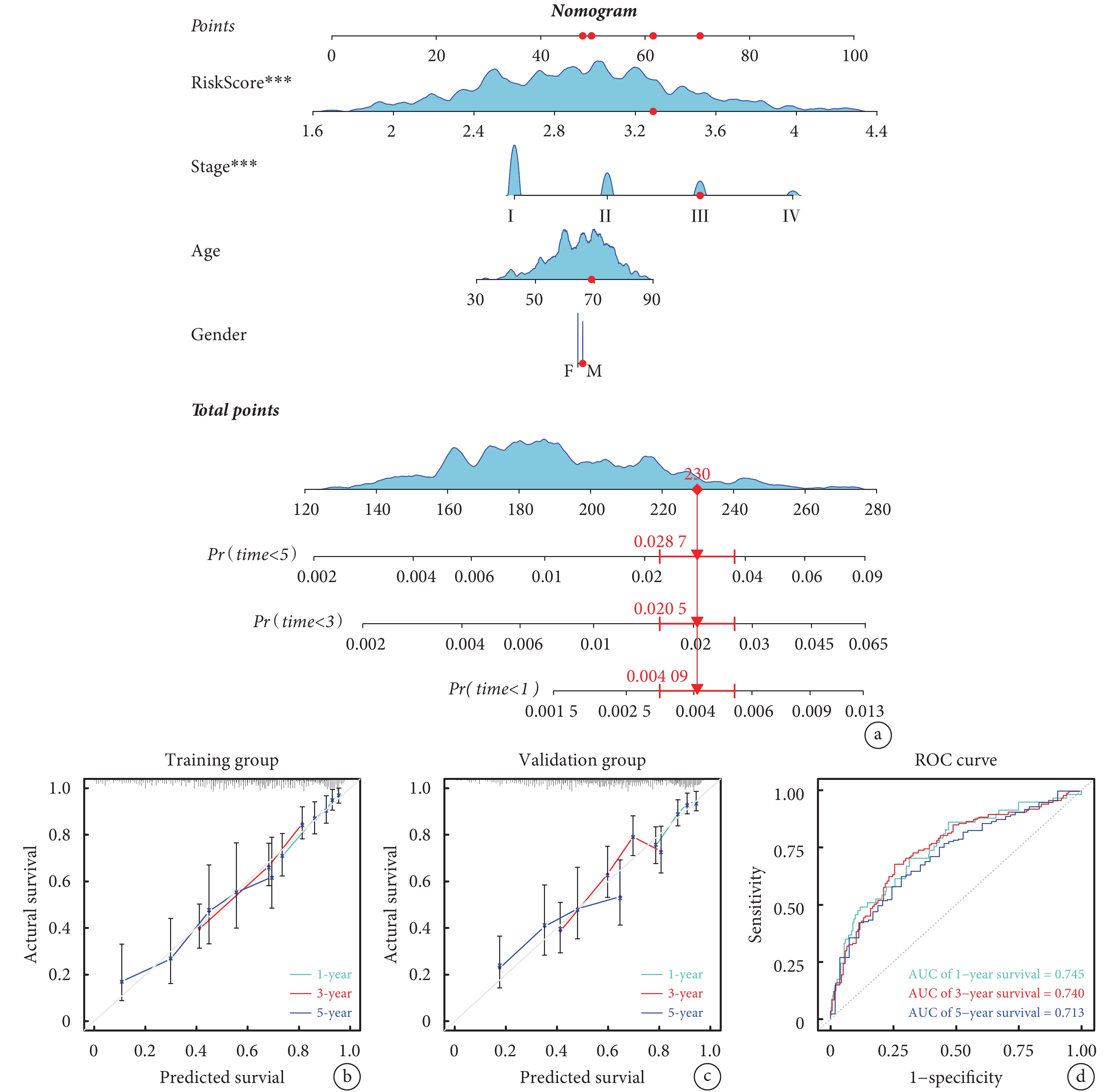 圖6
列線圖構建和診斷性能驗證
圖6
列線圖構建和診斷性能驗證
a:預測肺腺癌患者1、3、5年OS率的列線圖;b~c:分別為列線圖在訓練組和驗證組中預測1、3、5年OS率的校準曲線;d:列線圖預測1、3、5年OS率的ROC曲線;OS:總生存期;ROC:受試者工作特征
2.6 風險評分與免疫細胞浸潤的關系
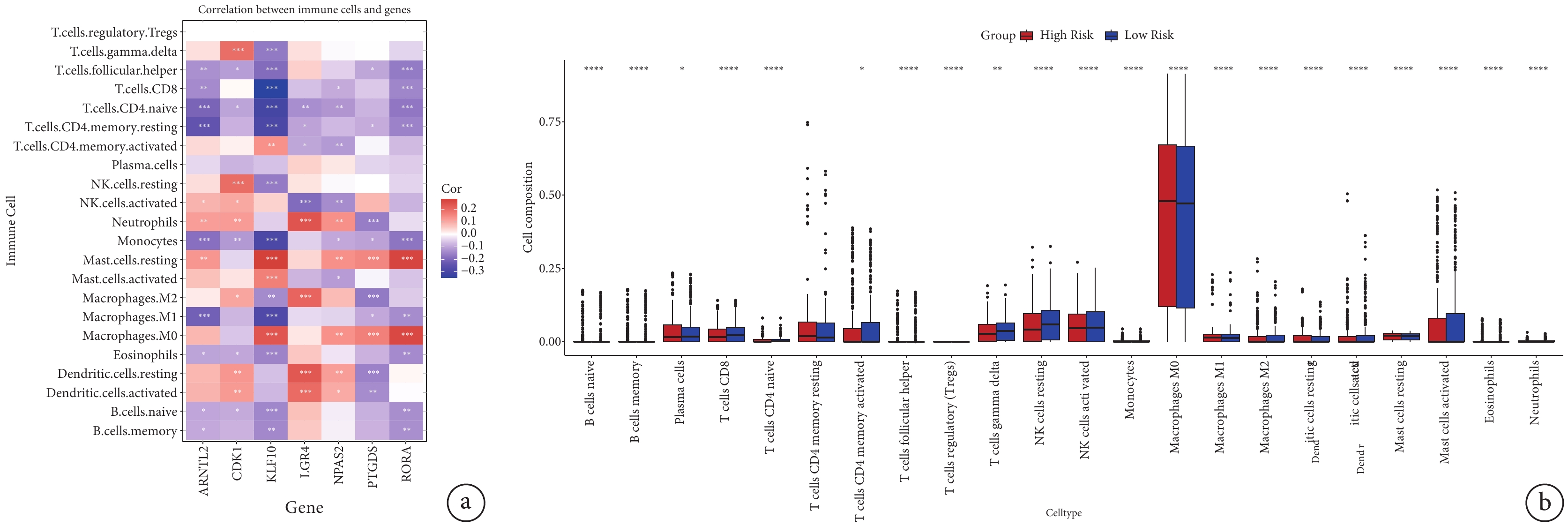
用CIBERSORT算法計算7個預后特征基因與22種不同類型免疫細胞之間的關系,評估RiskScore預測模型與免疫細胞浸潤之間的聯系。在本研究中,ARNTL2的表達主要與初始CD4 T細胞、靜息記憶CD4 T細胞、單核細胞和巨噬細胞的浸潤呈負相關;CDK1的表達主要與γδT細胞和靜息自然殺傷細胞的浸潤呈正相關;KLF10的表達主要與肥大細胞和M0巨噬細胞的浸潤呈正相關,與CD8 T細胞、初始CD4 T細胞、靜息記憶CD4 T細胞、單核細胞和M1巨噬細胞的浸潤呈負相關;LGR4的表達主要與中性粒細胞、巨噬細胞和樹突細胞的浸潤呈正相關;RORA的表達主要與靜息肥大細胞和M0巨噬細胞的浸潤呈正相關,與濾泡輔助性T細胞、CD4 T細胞、CD8 T細胞和單核細胞的浸潤呈負相關;見圖7a。此外,還比較了低風險組和高風險組的免疫細胞浸潤水平;見圖7b。在22種免疫細胞中,20種免疫細胞的浸潤比例在低風險組和高風險組之間存在顯著差異。特別是巨噬細胞,其在兩組中都占較高比例,并具有顯著差異,這表明控制該細胞的行為對干預LUAD患者的腫瘤進展是必不可少的。
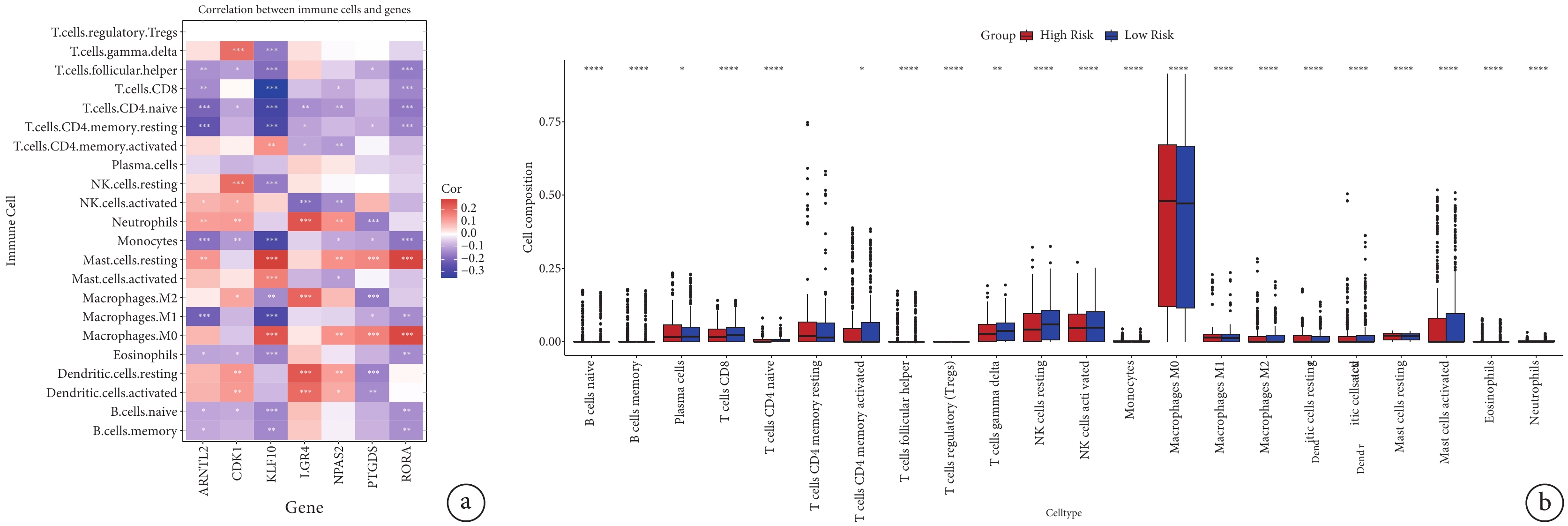 圖7
7個預后基因與免疫細胞浸潤的相關性
圖7
7個預后基因與免疫細胞浸潤的相關性
a:7個預后基因與22種免疫細胞浸潤的相關性;b:低風險組和高風險組腫瘤樣本中22種不同類型免疫細胞浸潤比例的差異
2.7 風險評分與免疫檢查點基因的相關性分析
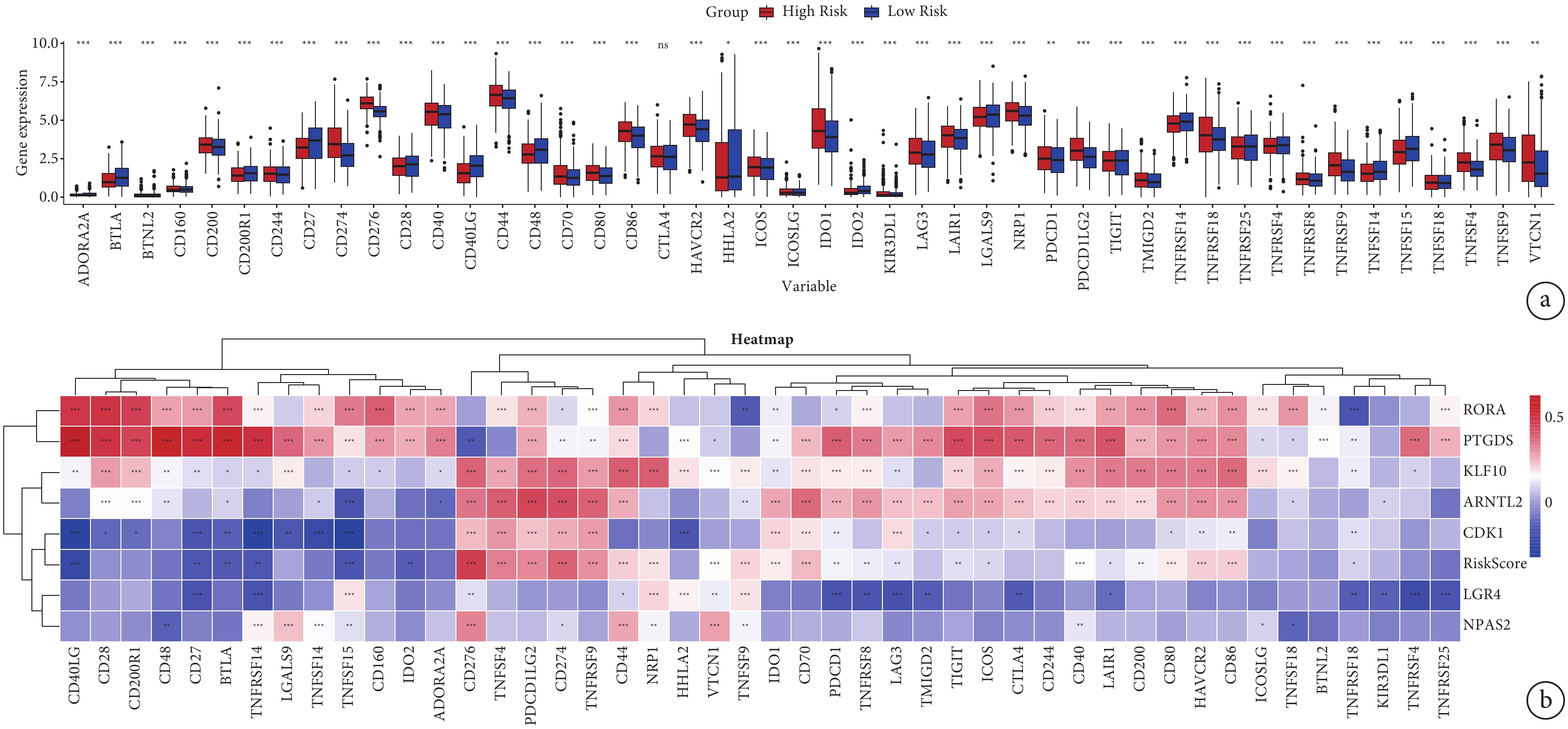
在LUAD數據集中,我們描述了RiskScore與46個常見免疫檢查點基因之間的相互作用。免疫檢查點基因在低風險組和高風險組的表達見圖8a。根據相關性分析,CD276、TNFSF4、PDCD1LG2、CD274和TNFRSF9等基因與RiskScore呈正相關,CD40LG和TNFSF15等基因與RiskScore呈負相關;見圖8b。上述結果可為尋找與免疫治療相關的生物靶點提供潛在方向。
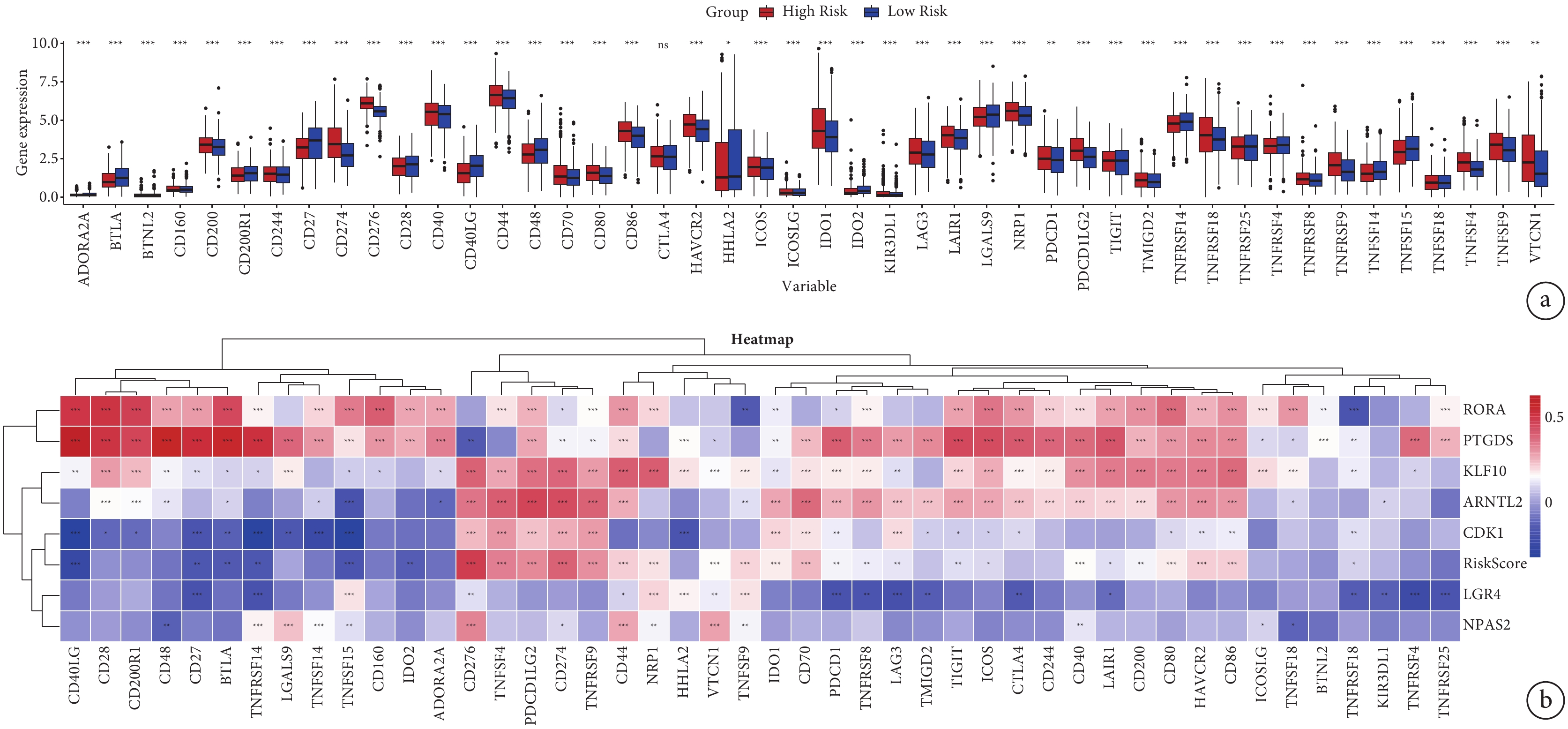 圖8
風險評分與46個常見免疫檢查點基因的相關性
圖8
風險評分與46個常見免疫檢查點基因的相關性
a:46個免疫檢查點基因在低風險組和高風險組的差異表達;b:風險評分與46個免疫檢查點基因的相關性
2.8 預后特征基因的表達與藥物敏感性的關系
經篩選得到860種藥物,將7個預后特征基因與藥物數據進行Pearson相關性分析,并計算7個預后特征基因表達量與藥物的敏感性。我們發現NPAS2與Refametinib、Trametinib和ARRY-162等藥物敏感性顯著相關(P<0.001);CDK1與Pyrazoloacridine的敏感性顯著相關(P<0.05);LGR4與Kahalide F、Dacarbazine和Ixazomib等藥物的敏感性顯著相關(P<0.010);KLF10與BLU-667和BMS-690514等藥物敏感性顯著相關(P<0.001);ARNTL2與Haloperidol的敏感性顯著相關(P<0.05)。這表明上述基因可作為這些藥物治療的生物靶點。
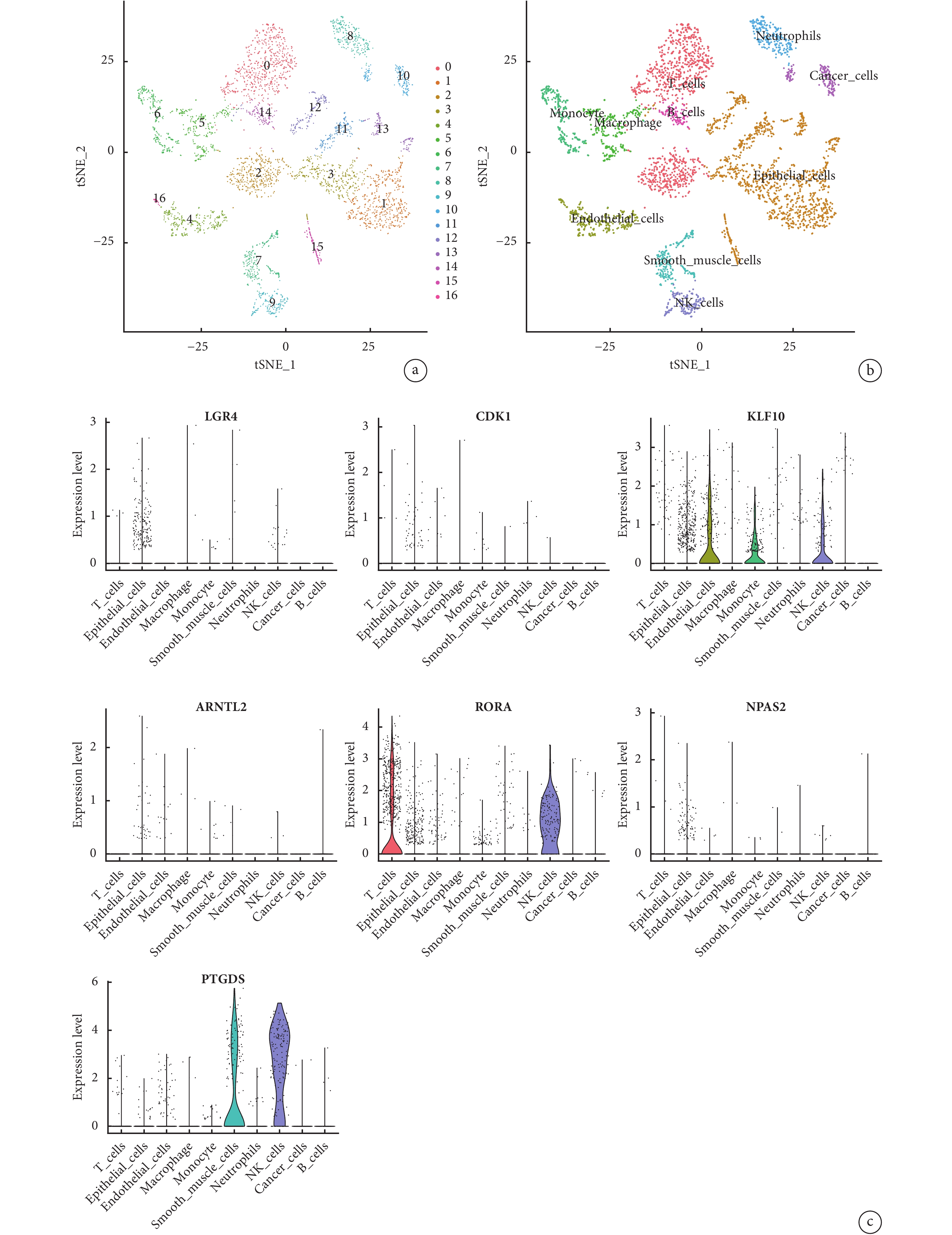
2.9 特征晝夜節律基因的單細胞轉錄分析
經過scRNA-seq數據處理和篩選,我們從2個LUAD樣本中獲得
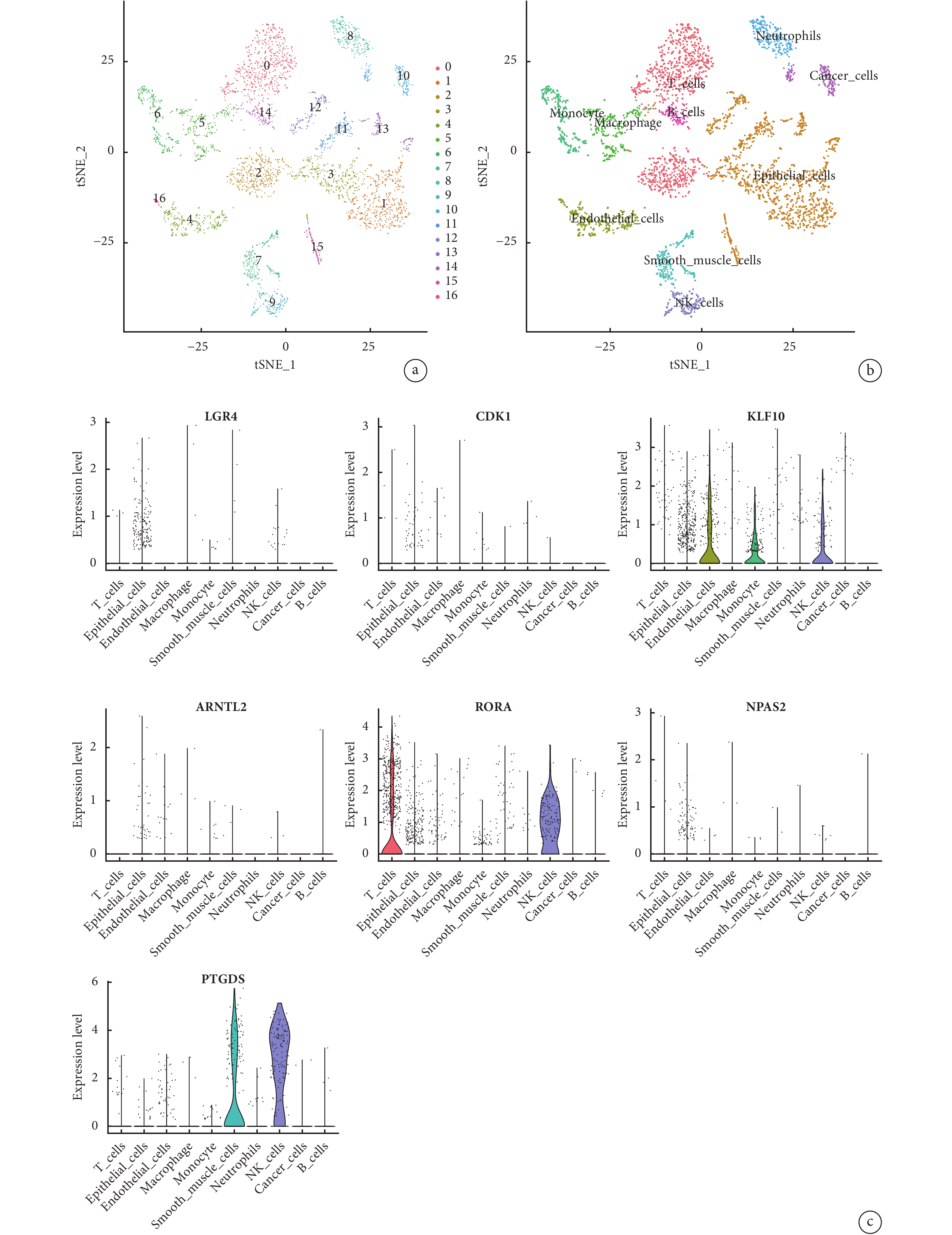 圖9
基于單細胞測序分析的7個晝夜節律基因表達譜
圖9
基于單細胞測序分析的7個晝夜節律基因表達譜
a:GSE149655單細胞的組成和分布;b:細胞標記基因鑒定細胞類型;c:7個晝夜節律基因的表達譜
3 討論
肺癌由于其高度侵襲性和預后差的特點,成為最致命的惡性腫瘤之一[14]。目前,LUAD的治療方式選擇主要根據組織學類型和臨床分期,但由于其高度異質性,即使是同樣組織學類型和臨床分期的LUAD患者預后也不相同[15]。現代醫學技術的進步極大提高了我們對LUAD致病機制的理解,并促進了新治療方法的發展,但仍缺乏針對早期LUAD有效的篩查和診斷措施[16]。人體晝夜節律影響正常生理活動,而晝夜節律紊亂也可能與肺癌的進展密切相關[17]。為進一步研究晝夜節律基因與LUAD的發生、發展、預后和TME之間的聯系,本研究篩選出特征晝夜節律基因,構建晝夜節律相關預后模型,同時結合scRNA-seq分析評估了晝夜節律紊亂與LUAD的關系,這一發現可能有助于開發新的LUAD治療策略。
首先,我們對LUAD遺傳變異數據進行了體細胞突變和CNVs分析,之后通過差異分析獲得57個晝夜節律DEGs,包括37個上調基因和20個下調基因,并對其進行了功能富集分析和PPI構建,經GO、KEGG和GSEA分析表明這些基因主要富集在晝夜節律行為、遺傳物質代謝調控、脂質代謝、AMPK、cGMP-PKG和JAK-STAT信號通路等相關生物學過程。研究[18-19]表明,脂代謝相關分子與晝夜節律基因的表達呈顯著正相關,并且增加了患者預后不良的風險。AMPK在腫瘤細胞代謝等調節過程中發揮重要作用。據文獻[20]報道,AMPK可通過直接調節自噬,靶向調節腫瘤細胞的代謝和微環境,進而影響腫瘤細胞的存活,表明AMPK在治療癌癥方面具有一定的應用潛力。研究[21]證明cGMP-PKG通路參與乳腺癌細胞的侵襲及凋亡等生物學過程。越來越多的證據[22-23]表明,JAK-STAT通路的過度激活與許多疾病的不良預后密切相關,包括黑色素瘤、膠質母細胞瘤、肺癌、乳腺癌、直腸癌和前列腺癌等。
其次,應用Cox回歸、LASSO回歸、SVM-RFE和隨機森林依次篩選57個DEGs,每種算法都有各自的優點。LASSO回歸分析主要用于過濾變量和避免模型過度擬合[24];SVM-RFE可方便地去除重復成分,在樣本較少的數據集中保留與結果相關的變量[25];隨機森林對候選基因進行排序、構建預測模型以及評估每個變量的相關性[26]。上述3種機器學習算法屬于集成特征的選擇,具有特定的優勢和要素篩選過程的可重復性,并且已經被證明在識別預后特征基因方面是有效的[27-28]。將每種方法得到的DEGs取交集,最終確定由7個預后特征晝夜節律基因(LGR4、CDK1、KLF10、ARNTL2、RORA、NPAS2、PTGDS)組成的RiskScore預測模型,根據中位RiskScore將樣本分為低風險組和高風險組。進一步對兩組患者的預后進行研究,結果顯示,高風險組患者的OS明顯短于低風險組,差異有統計學意義(P<0.001)。使用ROC曲線和時間依賴性ROC曲線表明模型具有良好的準確性,在外部驗證數據集中也得到驗證。然后用RiskScore和兩組具有獨立預后的臨床病理特征繪制出列線圖,該模型能精確預測患者1年、3、5年OS率,其AUC值分為0.745、0.740和0.713,校準曲線也顯示出準確的重疊。
最后,通過應用CIBERSORT算法評估免疫細胞在低風險組和高風險組的分布,我們發現巨噬細胞在兩組中都占較高比例。巨噬細胞是促炎細胞因子的主要來源之一,其生理活動主要受生物鐘控制[29]。實驗[30]表明晝夜節律紊亂會降低腫瘤中M1/M2巨噬細胞比例,促使TME免疫抑制,加速腫瘤生長,增加患者死亡率。根據相關性分析,KLF10和RORA的表達主要與CD8 T細胞、初始CD4 T細胞、靜息記憶CD4 T細胞、單核細胞和M1巨噬細胞的浸潤水平呈負相關;與靜息肥大細胞和M0巨噬細胞的浸潤水平呈正相關。此外,研究還發現,大多數免疫檢查點基因在高風險組中的表達水平較高,并且RiskScore與CD276、TNFSF4、PDCD1LG2、CD274和TNFRSF9的表達呈正相關。可見特征晝夜節律基因的表達與免疫細胞的浸潤水平息息相關。為此我們采用scRNA-seq技術,進一步分析了7個特征晝夜節律基因在不同免疫細胞群體間的分布及其分子特征。在本研究中,RORA和KLF10主要表達于自然殺傷細胞。上述分析結果進一步驗證了巨噬細胞和自然殺傷細胞等免疫細胞的活動一定程度上與晝夜節律相關,為將來從晝夜節律紊亂影響免疫細胞機制的角度尋找治療LUAD的潛在策略提供了可能的方向。
上述7個特征基因已被報道與多種癌癥的發生發展密切相關。LGR4是G蛋白偶聯受體(G protein-coupled receptors,GPCRs)超家族的跨膜受體成員之一,其與R-Spinins/Norrin配體結合,在Wnt介導的信號轉導中起關鍵作用[31]。越來越多的證據表明,LGR4在腫瘤組織中表達上調,并參與多種癌癥的發生、發展和轉移[32]。CDK1屬于絲氨酸/蘇氨酸蛋白激酶家族,是細胞周期G2期到M期過程中的一個檢查點,其表達與細胞進入或退出增殖周期直接相關[33]。已有研究[34]證實,CDK1可作為LUAD的預后指標,以及在LUAD的發生發展、細胞周期轉變和免疫相關通路治療反應中發揮重要作用。KLF10是SP(specificity proteins)/KLF(Kruppel-like factor)轉錄因子家族的一員,與細胞增殖、細胞凋亡和糖脂代謝等的重要聯系已逐漸被挖掘[35]。研究[36-37]表明,KLF10在人類致癌過程中起關鍵作用。ARNTL2作為一種晝夜節律轉錄因子,其在LUAD中高度表達,并促進癌細胞的增殖、遷移和侵襲,是LUAD患者預后不良的獨立預測因子[17]。RORA作為LUAD的核心時鐘控制基因,其已被證明與多種生物過程和疾病有關[38]。RORA有助于抗細胞凋亡和抗炎反應,抑制乳腺癌、前列腺癌和卵巢癌的細胞增殖[39]。此外,RORA在癌癥中經常失活,這使其成為癌癥治療的潛在靶點[40]。Du等[41]發現與肺癌組織樣本相比,非癌組織中RORA的表達水平升高。上述分析表明RORA可能是一種癌癥抑制因子,這與本研究結果一致。NPAS2被認為是腫瘤發生和免疫侵襲的重要調節因子,其表達增加導致肺癌患者的生存率降低[42-44]。相關研究[45]表明PTGDS在肺癌中表達下調并抑制腫瘤進展,是早期診斷和判斷預后的一個潛在腫瘤標志物。RORA和KLF10作為時鐘控制基因,其主要調節核心時鐘基因的表達[46]。在晝夜節律機制中,RORA與其他核受體REV-ERBα和REV-ERBβ(NR1D1和NR1D2)競爭,結合核心時鐘基因BMAL1(ARNTL1)啟動子中的特異性DNA反應元件,促進BMAL1的轉錄[47]。KLF10可調控多種晝夜節律基因的表達,是晝夜調節肝臟能量代謝等相關生物學過程所必需的[48]。
本研究證明了紊亂的晝夜節律與LUAD的免疫狀態有關。根據免疫細胞浸潤分析,高風險組樣本中巨噬細胞的浸潤率高于低風險組且在兩組中都占較高比例,scRNA-seq分析結果提示RORA和KLF10都主要表達于自然殺傷細胞。自然殺傷細胞是一種先天免疫細胞,其介導的免疫監測不僅能直接殺傷腫瘤細胞,還間接釋放細胞因子調節其他白細胞,如巨噬細胞、T細胞和樹突狀細胞[49]。研究[50]發現,晝夜節律紊亂不僅促進自然殺傷細胞凋亡,加速自然殺傷細胞衰老,而且也破壞自然殺傷細胞介導的免疫監視功能,表現為MHC-I缺陷的腫瘤細胞清除減少和B16黑色素瘤細胞的清除能力降低,從而促進腫瘤的發生發展。這說明TME中的免疫細胞普遍存在晝夜節律紊亂的現象,這為以后進一步從晝夜節律免疫機制的角度指導LUAD機制研究和藥物開發提供了理論支持。
綜上所述,本研究通過Cox回歸和3種不同的機器學習算法構建了準確有效的7個特征晝夜節律基因預后模型。基于該7個基因的RiskScore模型可預測LUAD患者的OS。將RiskScore和臨床參數相結合的列線圖可用于預測LUAD患者1、3、5年OS率,其有助于LUAD患者的預后和隨訪監測,為LUAD患者的個體化診療提供參考。但本研究仍存在一定的局限性。首先,我們的研究數據主要來自TCGA和GEO數據集,有必要在大型獨立臨床隊列中評估其預測效能。其次,我們缺乏深入的研究,特別是沒有設計基因組定向分層實驗。最后,這7個基因在LUAD發病中的生物學機制有待進一步通過功能研究來具體闡明。
利益沖突:無。
作者貢獻:崔嚴奇、趙虎和張亞偉負責病例篩選,數據整理與論文設計,初稿撰寫等;曾志勇、倪琳、連鐸煌、楊鯨蓉、葉仕新、許蜂蜂和張錦燦負責論文審閱與修改。
肺癌是全球癌癥相關死亡的主要原因[1]。非小細胞肺癌(non-small cell lung cancer,NSCLC)約占所有肺癌的85%,其主要組織學亞型是肺腺癌(lung adenocarcinoma,LUAD),約占肺癌發病率的40%[2]。盡管LUAD的靶向治療和免疫治療取得了重大進展,但由于疾病的隱匿性和缺乏特異性,大多數患者在確診時已處于晚期,5年總生存率仍低于20%[3]。因此,迫切需要探索新的生物標志物和可靠的預后預測模型來改善LUAD患者的生存結局。
晝夜節律系統協調人體生理和行為節律以適應24 h生理周期[4]。晝夜節律基因主要分為兩類:核心時鐘基因和時鐘控制基因,前者控制人體生理晝夜節律,后者調節核心時鐘基因的表達[5]。許多研究[4,6-7]表明,晝夜節律紊亂不僅會導致代謝、心血管和免疫功能障礙,而且還與患癌風險和較差的預后有關。隨著現代醫學的發展,在高通量RNA測序技術和人工智能的輔助下,生物信息學結合機器學習算法已被廣泛應用于基因組和蛋白質組研究,越來越多疾病的篩查、診斷和預后模型被開發出來[8]。單細胞RNA測序技術(single cell sequencing,scRNA-seq)被用于分析腫瘤微環境(tumor microenvironment,TME)的細胞類型和腫瘤異質性等相關生物學信息[9],能夠在單細胞水平上檢測基因表達量,從而有助于剖析腫瘤細胞群體中基因表達的異質性[10]。借助這一優勢,scRNA-seq和生物信息學數據分析方法的發展為揭示TME中不同細胞群體的分子特征提供了前所未有的機會[3]。
1 資料與方法
本研究首先對癌癥基因組圖譜(The Cancer Genome Atlas,TCGA)的LUAD晝夜節律基因表達量和臨床病理數據進行全面分析。然后通過Cox回歸和3種不同的機器學習算法篩選出與LUAD預后相關的特征晝夜節律基因,構建LUAD預后的晝夜節律基因預測模型,并驗證該模型在外部數據集的穩定性。此外,我們還評估了該模型與免疫細胞和免疫檢查點基因的關聯。最后在scRNA-seq分析的基礎上,探討預后相關的特征晝夜節律基因與TME中不同免疫細胞群體之間的分子特征。研究流程見圖1。
圖1
研究流程圖
1.1 數據來源
晝夜節律基因集(210個基因)于2023年6月27日從晝夜節律基因數據庫(Circadian Genes Database,CGDB;http://cgdb.biocuckoo.org)得到,同期在TCGA數據庫(https://portal.gdc.cancer.gov)獲得LUAD RNA-Seq數據和臨床病理數據(524例癌癥樣本,58例癌旁正常組織樣本),篩選出LUAD患者晝夜節律基因表達量,并經過log2的轉化。從Gene Expression Omnibus(GEO)數據庫(https://www.ncbi.nlm.nih.gov/geo)獲得外部驗證數據集GSE68465(443例癌癥樣本,19例癌旁正常組織樣本)。
1.2 晝夜節律差異基因篩選、突變及拷貝數變異分析
從TCGA數據庫下載LUAD基因突變數據和拷貝數變異(copy number variations,CNVs)數據,與晝夜節律基因相對應,分別通過R-4.3.0軟件“maftools”和“Rcircos”包處理并可視化。采用“DESeq2”包以P≤0.05和|log2(fold change)|>1的標準篩選晝夜節律差異表達基因(differentially expressed genes,DEGs),并使用“pheatmap”“ggplot2”和“psych”包繪制火山圖和基因與臨床性狀相關性熱圖。
1.3 功能富集分析和構建蛋白質相互作用網絡
使用“enrichplot”“org.Hs.eg.db”和“clusterProfiler”包對DEGs進行基因本體功能富集分析(gene ontology,GO)、京都基因與基因組百科全書通路分析(Kyoto Encyclopedia of Genes and Genomes,KEGG)和基因集富集分析(gene set enrichment analysis,GSEA),使用“ggplot2”包對結果進行可視化。通過The STRING網站(www.string-db.org)以最低相關分數0.400為閾值生成DEGs蛋白質相互作用網絡(protein-protein interaction network,PPI)[11]。數據以TSV格式從數據庫下載,并用Cytoscape_v3.9.0軟件可視化。P≤0.05為差異有統計學意義。
1.4 構建風險評分預測模型
將訓練組524例LUAD樣本的晝夜節律DEGs表達量和生存信息合并,剔除15例無總生存期(overall survival,OS)及生存狀態的患者,余509例LUAD樣本依次納入Cox回歸和3種機器學習算法[最小絕對收縮和選擇算子(LASSO)回歸、支持向量機遞歸特征消除(SVM-RFE)、隨機森林]。選擇4種算法計算DEGs的交集基因,最終構建風險評分(RiskScore)預測模型。RiskScore預測模型計算公式:RiskScore=Exp1×C1+Exp2×C2+…+Expn×Cn(Exp為預后特征基因的表達量,C為LASSO回歸分析得到的回歸系數,n為交集基因的數目)。
根據上面的公式計算每例樣本的RiskScore,以RiskScore中位數為截斷值將LUAD樣本分為低風險組和高風險組,P≤0.05為差異有統計學意義。
1.5 評價風險預測模型和繪制列線圖
首先繪制Kaplan-Meier生存曲線比較低風險組和高風險組的OS。其次通過“timeROC”包繪制時間依賴性受試者工作特征(receiver operating characteristic,ROC)曲線,計算出訓練組中模型預測1、3、5年OS率的曲線下面積(area under the curve,AUC),評價預測風險模型的準確性。最后將兩組的臨床病理因素和RiskScore納入Cox回歸分析,并構建列線圖。利用校準曲線和ROC曲線評價生存預測的準確性。使用GSE68465數據集驗證構建的預測模型。
1.6 評估腫瘤微環境的免疫學特征
通過應用“CIBERSORT”包對22種不同免疫細胞的浸潤狀態進行評估,以探討RiskScore與免疫細胞浸潤之間的關系。此外,從相關文獻中檢索出46個常見的免疫檢查點,探索RiskScore與46個免疫檢查點基因之間的聯系[12]。P≤0.05為差異有統計學意義。
1.7 藥物敏感性分析
相關基因表達和藥物數據從CellMiner數據庫(https://discover.nci.nih.gov/cellminer/home.do)下載。使用臨床實驗室驗證以及國家藥品監督管理局標準認證相結合的方法對藥物數據進行篩選。然后,將預后特征晝夜節律基因的表達數據與藥物數據合并,進行Pearson相關性檢驗,確定其相關性和藥物敏感性。
1.8 特征晝夜節律基因的單細胞群體分子特征
為進一步驗證特征晝夜節律基因與TME的關系,采用來自GSE149655數據集(GSM4506699、GSM4506701)的2例LUAD 10×scRNA-seq數據,并利用“Seurat”“patchwork”和“dplyr”包進行數據整理、標準化以及主成分分析(principal component analysis,PCA)。首先通過以下篩選標準保留高質量scRNA-seq數據:(1)剔除線粒體基因超過25%的細胞;(2)提取表達>200個且<
2 結果
2.1 晝夜節律基因的差異表達和遺傳變異模式
遺傳變異數據與晝夜節律基因一一對應后得到的基因突變和CNVs數據(201個基因,447例樣本)在LUAD中的體細胞突變頻率為68.01%(304/447);見圖2a。從TCGA數據庫收集的LUAD患者晝夜節律基因以P≤0.05和|log2(fold change)|>1為閾值,篩選出57個DEGs,包括37個上調基因和20個下調基因,晝夜節律DEGs在染色體的位置見圖2b。DEGs與臨床性狀相關性熱圖、DEGs的火山圖可視化結果見圖2c~d。
圖2
肺腺癌晝夜節律基因的分子特征和表達變異
a:447 例肺腺癌患者晝夜節律基因的突變頻率,每列代表 1 例患者,頂部的條形圖代表腫瘤突變負荷,右側的數字代表每個基因的突變頻率,右側的條形圖顯示了每種基因的比例,下面堆疊的條形圖顯示了每個樣本中轉化的百分比;b:差異表達基因在23條染色體上的位置;c:差異表達基因與臨床特征的相關性熱圖;d:晝夜節律差異表達基因火山圖
2.2 功能富集分析和差異表達基因蛋白質相互作用網絡的構建
為探索LUAD中晝夜節律基因的相關生物學功能和途徑,對上述57個DEGs進行GO及KEGG分析,富集度最高的GO分類和KEGG通路見圖3a~b。GO分析顯示,DEGs主要富集在晝夜節律行為、RNA代謝的負調控、序列特異性DNA結合等相關生物學功能;KEGG分析顯示,DEGs主要富集在AMPK信號通路、脂肪細胞脂解的調控通路和神經活性配體-受體相互作用通路等。將P≤0.05的晝夜節律基因納入GSEA分析,其主要富集于cGMP-PKG信號通路、脂質與動脈粥樣硬化和JAK-STAT信號通路等相關生物學過程,進一步驗證了遺傳物質調控和脂肪酸等能量物質代謝與腫瘤進展的密切關系。
圖3
功能富集分析和 PPI 網絡的構建
a:GO 分析,對前 10 個富集類別(生物過程、細胞成分和分子功能)進行可視化;b:KEGG 分析,顯示前 9 個富集通路;c:PPI 網絡構建,基于 PPI 網絡的前 46 個節點差異表達基因,使用 Degree 算法選擇基因,顏色越深表示基因關聯的節點越多;PPI:蛋白質相互作用網絡
在去除不與其他節點交互的DEGs后,通過Degree算法構建了46個節點的PPI網絡,展示晝夜節律DEGs蛋白質之間的相互作用;見圖3c[13]。以上分析結果解釋了基因層面的關系,也為將來尋找與預后相關生物學標志物的潛在靶點提供了研究思路。
2.3 篩選預后特征基因并構建風險評分預測模型
將上述57個DEGs分別納入Cox回歸、LASSO回歸、SVM-RFE和隨機森林。Cox回歸得到17個與預后獨立相關的基因(P<0.05);見圖4a。通過LASSO回歸的降維分析,獲得31個特征基因;見圖4b~c。應用SVM-RFE方法根據其重要性評分鑒定出25個準確率最高的基因;見圖4d。通過隨機森林算法對DEGs重要性評分排序后,識別出25個候選基因;見圖4e。最后,將每種方法篩選的DEGs取交集后的7個預后相關特征基因(LGR4、CDK1、KLF10、ARNTL2、RORA、NPAS2、PTGDS)可視化為Venn圖;見圖4f。根據7個基因的表達量和風險系數構建RiskScore模型:RiskScore=LGR4×
圖4
機器學習算法篩選生物標志物
a:Cox 分析顯示 17 個基因與預后顯著相關;b~c:LASSO 回歸分析用于篩選晝夜節律基因的生物標志物,虛線標注處為Log(
2.4 風險評分與臨床病理特征的關系
以訓練組LUAD樣本的RiskScore中位數為截斷值將訓練組樣本分為低風險組(n=255)和高風險組(n=254)。不同風險組患者表現出不同的臨床病理特征。病理分期(P<0.001)、T分期(P=0.02)和N分期(P<0.001)均與RiskScore相關,然而年齡(P=0.59)和性別(P=0.31)與RiskScore無關。LGR4(R=0.419,P<0.001)、CDK1(R=0.710,P<0.001)、KLF10(R=0.499,P<0.001)、ARNTL2(R=0.736,P<0.001)和NPAS2(R=0.523,P<0.001)與RiskScore呈正相關,RORA(R=?0.204,P<0.001)和PTGDS(R=?0.356,P<0.001)與RiskScore呈負相關。
2.5 檢驗風險評分預測模型性能并繪制列線圖
Kaplan-Meier生存曲線顯示,訓練組樣本中,高風險和低風險組OS差異有統計學意義(P<0.001),表明高風險組比低風險組預后差;見圖5a。為確保RiskScore預測模型的穩定性,繪制ROC曲線和時間依賴性ROC曲線,ROC曲線的AUC值為0.643,時間依賴性ROC曲線顯示,預測1、3、5年OS率的AUC值分別為0.702、0.680、0.654,表明模型具有良好的準確性;見圖5b~c。驗證組數據集剔除1例無OS的樣本后,將剩余442例樣本以相同方法分為低風險組(n=221)和高風險組(n=221),然后繪制Kaplan-Meier生存曲線、ROC曲線及時間依賴性ROC曲線,兩組OS差異仍有統計學意義(P<0.001)。ROC曲線的AUC值為0.643,時間依賴性ROC曲線顯示,預測1、3、5年OS率的AUC值分別為0.730、0.688、0.653;見圖5d~f。上述結果說明該模型在驗證組中仍具有較好的預測性能。
圖5
評估RiskScore模型的預測價值
a~c:分別為訓練組的Kaplan-Meier生存曲線、ROC曲線和時間依賴性ROC曲線;d~f:分別為驗證組的Kaplan-Meier生存曲線、ROC曲線和時間依賴性ROC曲線;ROC:受試者工作特征;AUC:曲線下面積
通過評估基于晝夜節律基因的風險模型在臨床實踐中的有效性,將RiskScore及臨床病理特征納入Cox回歸分析,篩選LUAD患者OS的獨立危險因素,并構建列線圖;見圖6a。該個體化預測模型可預測LUAD患者1、3、5年OS率,其AUC值分別為0.745、0.740和0.713。校準曲線和ROC曲線顯示,列線圖預測結果可靠;見圖6b~d。
圖6
列線圖構建和診斷性能驗證
a:預測肺腺癌患者1、3、5年OS率的列線圖;b~c:分別為列線圖在訓練組和驗證組中預測1、3、5年OS率的校準曲線;d:列線圖預測1、3、5年OS率的ROC曲線;OS:總生存期;ROC:受試者工作特征
2.6 風險評分與免疫細胞浸潤的關系
用CIBERSORT算法計算7個預后特征基因與22種不同類型免疫細胞之間的關系,評估RiskScore預測模型與免疫細胞浸潤之間的聯系。在本研究中,ARNTL2的表達主要與初始CD4 T細胞、靜息記憶CD4 T細胞、單核細胞和巨噬細胞的浸潤呈負相關;CDK1的表達主要與γδT細胞和靜息自然殺傷細胞的浸潤呈正相關;KLF10的表達主要與肥大細胞和M0巨噬細胞的浸潤呈正相關,與CD8 T細胞、初始CD4 T細胞、靜息記憶CD4 T細胞、單核細胞和M1巨噬細胞的浸潤呈負相關;LGR4的表達主要與中性粒細胞、巨噬細胞和樹突細胞的浸潤呈正相關;RORA的表達主要與靜息肥大細胞和M0巨噬細胞的浸潤呈正相關,與濾泡輔助性T細胞、CD4 T細胞、CD8 T細胞和單核細胞的浸潤呈負相關;見圖7a。此外,還比較了低風險組和高風險組的免疫細胞浸潤水平;見圖7b。在22種免疫細胞中,20種免疫細胞的浸潤比例在低風險組和高風險組之間存在顯著差異。特別是巨噬細胞,其在兩組中都占較高比例,并具有顯著差異,這表明控制該細胞的行為對干預LUAD患者的腫瘤進展是必不可少的。
圖7
7個預后基因與免疫細胞浸潤的相關性
a:7個預后基因與22種免疫細胞浸潤的相關性;b:低風險組和高風險組腫瘤樣本中22種不同類型免疫細胞浸潤比例的差異
2.7 風險評分與免疫檢查點基因的相關性分析
在LUAD數據集中,我們描述了RiskScore與46個常見免疫檢查點基因之間的相互作用。免疫檢查點基因在低風險組和高風險組的表達見圖8a。根據相關性分析,CD276、TNFSF4、PDCD1LG2、CD274和TNFRSF9等基因與RiskScore呈正相關,CD40LG和TNFSF15等基因與RiskScore呈負相關;見圖8b。上述結果可為尋找與免疫治療相關的生物靶點提供潛在方向。
圖8
風險評分與46個常見免疫檢查點基因的相關性
a:46個免疫檢查點基因在低風險組和高風險組的差異表達;b:風險評分與46個免疫檢查點基因的相關性
2.8 預后特征基因的表達與藥物敏感性的關系
經篩選得到860種藥物,將7個預后特征基因與藥物數據進行Pearson相關性分析,并計算7個預后特征基因表達量與藥物的敏感性。我們發現NPAS2與Refametinib、Trametinib和ARRY-162等藥物敏感性顯著相關(P<0.001);CDK1與Pyrazoloacridine的敏感性顯著相關(P<0.05);LGR4與Kahalide F、Dacarbazine和Ixazomib等藥物的敏感性顯著相關(P<0.010);KLF10與BLU-667和BMS-690514等藥物敏感性顯著相關(P<0.001);ARNTL2與Haloperidol的敏感性顯著相關(P<0.05)。這表明上述基因可作為這些藥物治療的生物靶點。
2.9 特征晝夜節律基因的單細胞轉錄分析
經過scRNA-seq數據處理和篩選,我們從2個LUAD樣本中獲得
圖9
基于單細胞測序分析的7個晝夜節律基因表達譜
a:GSE149655單細胞的組成和分布;b:細胞標記基因鑒定細胞類型;c:7個晝夜節律基因的表達譜
3 討論
肺癌由于其高度侵襲性和預后差的特點,成為最致命的惡性腫瘤之一[14]。目前,LUAD的治療方式選擇主要根據組織學類型和臨床分期,但由于其高度異質性,即使是同樣組織學類型和臨床分期的LUAD患者預后也不相同[15]。現代醫學技術的進步極大提高了我們對LUAD致病機制的理解,并促進了新治療方法的發展,但仍缺乏針對早期LUAD有效的篩查和診斷措施[16]。人體晝夜節律影響正常生理活動,而晝夜節律紊亂也可能與肺癌的進展密切相關[17]。為進一步研究晝夜節律基因與LUAD的發生、發展、預后和TME之間的聯系,本研究篩選出特征晝夜節律基因,構建晝夜節律相關預后模型,同時結合scRNA-seq分析評估了晝夜節律紊亂與LUAD的關系,這一發現可能有助于開發新的LUAD治療策略。
首先,我們對LUAD遺傳變異數據進行了體細胞突變和CNVs分析,之后通過差異分析獲得57個晝夜節律DEGs,包括37個上調基因和20個下調基因,并對其進行了功能富集分析和PPI構建,經GO、KEGG和GSEA分析表明這些基因主要富集在晝夜節律行為、遺傳物質代謝調控、脂質代謝、AMPK、cGMP-PKG和JAK-STAT信號通路等相關生物學過程。研究[18-19]表明,脂代謝相關分子與晝夜節律基因的表達呈顯著正相關,并且增加了患者預后不良的風險。AMPK在腫瘤細胞代謝等調節過程中發揮重要作用。據文獻[20]報道,AMPK可通過直接調節自噬,靶向調節腫瘤細胞的代謝和微環境,進而影響腫瘤細胞的存活,表明AMPK在治療癌癥方面具有一定的應用潛力。研究[21]證明cGMP-PKG通路參與乳腺癌細胞的侵襲及凋亡等生物學過程。越來越多的證據[22-23]表明,JAK-STAT通路的過度激活與許多疾病的不良預后密切相關,包括黑色素瘤、膠質母細胞瘤、肺癌、乳腺癌、直腸癌和前列腺癌等。
其次,應用Cox回歸、LASSO回歸、SVM-RFE和隨機森林依次篩選57個DEGs,每種算法都有各自的優點。LASSO回歸分析主要用于過濾變量和避免模型過度擬合[24];SVM-RFE可方便地去除重復成分,在樣本較少的數據集中保留與結果相關的變量[25];隨機森林對候選基因進行排序、構建預測模型以及評估每個變量的相關性[26]。上述3種機器學習算法屬于集成特征的選擇,具有特定的優勢和要素篩選過程的可重復性,并且已經被證明在識別預后特征基因方面是有效的[27-28]。將每種方法得到的DEGs取交集,最終確定由7個預后特征晝夜節律基因(LGR4、CDK1、KLF10、ARNTL2、RORA、NPAS2、PTGDS)組成的RiskScore預測模型,根據中位RiskScore將樣本分為低風險組和高風險組。進一步對兩組患者的預后進行研究,結果顯示,高風險組患者的OS明顯短于低風險組,差異有統計學意義(P<0.001)。使用ROC曲線和時間依賴性ROC曲線表明模型具有良好的準確性,在外部驗證數據集中也得到驗證。然后用RiskScore和兩組具有獨立預后的臨床病理特征繪制出列線圖,該模型能精確預測患者1年、3、5年OS率,其AUC值分為0.745、0.740和0.713,校準曲線也顯示出準確的重疊。
最后,通過應用CIBERSORT算法評估免疫細胞在低風險組和高風險組的分布,我們發現巨噬細胞在兩組中都占較高比例。巨噬細胞是促炎細胞因子的主要來源之一,其生理活動主要受生物鐘控制[29]。實驗[30]表明晝夜節律紊亂會降低腫瘤中M1/M2巨噬細胞比例,促使TME免疫抑制,加速腫瘤生長,增加患者死亡率。根據相關性分析,KLF10和RORA的表達主要與CD8 T細胞、初始CD4 T細胞、靜息記憶CD4 T細胞、單核細胞和M1巨噬細胞的浸潤水平呈負相關;與靜息肥大細胞和M0巨噬細胞的浸潤水平呈正相關。此外,研究還發現,大多數免疫檢查點基因在高風險組中的表達水平較高,并且RiskScore與CD276、TNFSF4、PDCD1LG2、CD274和TNFRSF9的表達呈正相關。可見特征晝夜節律基因的表達與免疫細胞的浸潤水平息息相關。為此我們采用scRNA-seq技術,進一步分析了7個特征晝夜節律基因在不同免疫細胞群體間的分布及其分子特征。在本研究中,RORA和KLF10主要表達于自然殺傷細胞。上述分析結果進一步驗證了巨噬細胞和自然殺傷細胞等免疫細胞的活動一定程度上與晝夜節律相關,為將來從晝夜節律紊亂影響免疫細胞機制的角度尋找治療LUAD的潛在策略提供了可能的方向。
上述7個特征基因已被報道與多種癌癥的發生發展密切相關。LGR4是G蛋白偶聯受體(G protein-coupled receptors,GPCRs)超家族的跨膜受體成員之一,其與R-Spinins/Norrin配體結合,在Wnt介導的信號轉導中起關鍵作用[31]。越來越多的證據表明,LGR4在腫瘤組織中表達上調,并參與多種癌癥的發生、發展和轉移[32]。CDK1屬于絲氨酸/蘇氨酸蛋白激酶家族,是細胞周期G2期到M期過程中的一個檢查點,其表達與細胞進入或退出增殖周期直接相關[33]。已有研究[34]證實,CDK1可作為LUAD的預后指標,以及在LUAD的發生發展、細胞周期轉變和免疫相關通路治療反應中發揮重要作用。KLF10是SP(specificity proteins)/KLF(Kruppel-like factor)轉錄因子家族的一員,與細胞增殖、細胞凋亡和糖脂代謝等的重要聯系已逐漸被挖掘[35]。研究[36-37]表明,KLF10在人類致癌過程中起關鍵作用。ARNTL2作為一種晝夜節律轉錄因子,其在LUAD中高度表達,并促進癌細胞的增殖、遷移和侵襲,是LUAD患者預后不良的獨立預測因子[17]。RORA作為LUAD的核心時鐘控制基因,其已被證明與多種生物過程和疾病有關[38]。RORA有助于抗細胞凋亡和抗炎反應,抑制乳腺癌、前列腺癌和卵巢癌的細胞增殖[39]。此外,RORA在癌癥中經常失活,這使其成為癌癥治療的潛在靶點[40]。Du等[41]發現與肺癌組織樣本相比,非癌組織中RORA的表達水平升高。上述分析表明RORA可能是一種癌癥抑制因子,這與本研究結果一致。NPAS2被認為是腫瘤發生和免疫侵襲的重要調節因子,其表達增加導致肺癌患者的生存率降低[42-44]。相關研究[45]表明PTGDS在肺癌中表達下調并抑制腫瘤進展,是早期診斷和判斷預后的一個潛在腫瘤標志物。RORA和KLF10作為時鐘控制基因,其主要調節核心時鐘基因的表達[46]。在晝夜節律機制中,RORA與其他核受體REV-ERBα和REV-ERBβ(NR1D1和NR1D2)競爭,結合核心時鐘基因BMAL1(ARNTL1)啟動子中的特異性DNA反應元件,促進BMAL1的轉錄[47]。KLF10可調控多種晝夜節律基因的表達,是晝夜調節肝臟能量代謝等相關生物學過程所必需的[48]。
本研究證明了紊亂的晝夜節律與LUAD的免疫狀態有關。根據免疫細胞浸潤分析,高風險組樣本中巨噬細胞的浸潤率高于低風險組且在兩組中都占較高比例,scRNA-seq分析結果提示RORA和KLF10都主要表達于自然殺傷細胞。自然殺傷細胞是一種先天免疫細胞,其介導的免疫監測不僅能直接殺傷腫瘤細胞,還間接釋放細胞因子調節其他白細胞,如巨噬細胞、T細胞和樹突狀細胞[49]。研究[50]發現,晝夜節律紊亂不僅促進自然殺傷細胞凋亡,加速自然殺傷細胞衰老,而且也破壞自然殺傷細胞介導的免疫監視功能,表現為MHC-I缺陷的腫瘤細胞清除減少和B16黑色素瘤細胞的清除能力降低,從而促進腫瘤的發生發展。這說明TME中的免疫細胞普遍存在晝夜節律紊亂的現象,這為以后進一步從晝夜節律免疫機制的角度指導LUAD機制研究和藥物開發提供了理論支持。
綜上所述,本研究通過Cox回歸和3種不同的機器學習算法構建了準確有效的7個特征晝夜節律基因預后模型。基于該7個基因的RiskScore模型可預測LUAD患者的OS。將RiskScore和臨床參數相結合的列線圖可用于預測LUAD患者1、3、5年OS率,其有助于LUAD患者的預后和隨訪監測,為LUAD患者的個體化診療提供參考。但本研究仍存在一定的局限性。首先,我們的研究數據主要來自TCGA和GEO數據集,有必要在大型獨立臨床隊列中評估其預測效能。其次,我們缺乏深入的研究,特別是沒有設計基因組定向分層實驗。最后,這7個基因在LUAD發病中的生物學機制有待進一步通過功能研究來具體闡明。
利益沖突:無。
作者貢獻:崔嚴奇、趙虎和張亞偉負責病例篩選,數據整理與論文設計,初稿撰寫等;曾志勇、倪琳、連鐸煌、楊鯨蓉、葉仕新、許蜂蜂和張錦燦負責論文審閱與修改。