

食管癌(esophageal cancer,EC)是第8大常見癌癥,也是全球癌癥死亡的第6大原因,嚴重影響全球人類健康[1]。食管癌的5年生存率很低,約為12%~20%[2]。吸煙和飲酒是食管癌的兩大危險因素[3-6]。此外,遺傳易感性、體質量指數增加、必需微量營養素攝入、口腔感染和飲食改變等因素也是食管癌高發的誘因[7-10]。目前,食管癌的治療采用包括手術、化學治療、放射治療和免疫靶向治療在內的綜合治療為主[11],但針對食管癌病因的干預方式較少,及時針對病因進行干預降低食管癌發生率,有助于改善食管癌患者的生存。
腸道菌群與人體新陳代謝、免疫調節和神經內分泌系統密切相關[12]。健康人90% 以上的細菌來自厚壁菌門和擬桿菌門,其次是疣微菌門、變形菌門和放線菌門,共占微生物的99%[13]。越來越多的證據表明,食管癌患者的特定細菌和細菌菌群失調,可通過破壞DNA、激活致癌信號通路、產生促瘤代謝物和抑制抗腫瘤免疫來促進食管癌進展[14]。Zhou等[15]發現,高豐度的產酸細菌(主要是乳酸桿菌、雙歧桿菌、葡萄球菌和鏈球菌)可通過乳酸代謝失調促進致癌作用。Münch等[16]使用Barrett食管的動物模型發現,高脂肪飲食誘導的腸道微生物群變化導致促炎細胞因子和免疫細胞水平升高,繼而導致促腫瘤免疫表型。腸道微生物可起保護作用,其擾動和水平降低能促進炎性反應和縮短生存時間[17]。根據這些研究,塑造腸道微生物群的組成可能有望實現有效和特異性的抗腫瘤免疫反應。基于此,我們從孟德爾遺傳角度出發,對腸道菌群和食管癌的關系進行研究。
與觀察性研究不同,孟德爾隨機化(Mendelian randomization,MR)分析是使用遺傳變異作為工具變量(instrumentalvariables,IVs)來推斷暴露與結果之間的因果關系,遺傳變異和結局之間不受混雜因素的影響,可作為研究腸道微生物與食管癌之間因果關系的一種新方法[18]。MR已被廣泛用于研究腸道微生物與疾病之間的關聯,包括癌癥[19]、免疫性疾病[20]、代謝性疾病[21]。在該研究中,我們使用全基因組關聯研究(genome-wide association studies,GWAS)的匯總統計數據進行雙向MR分析,以評估腸道菌群和食管癌的因果關系。
1 資料與方法
1.1 研究設計
本研究將腸道菌群作為暴露因素,選取顯著相關的單核苷酸多態性(single-nucleotide polymorphism,SNP)作為IVs,食管癌作為結局變量。基于公開的GWAS匯總統計數據,使用雙樣本雙向MR分析方法評估196個細菌分類群與食管癌之間的因果關系。使用異質性檢驗和水平多效性檢驗等分析方法驗證結果的可靠性和穩定性。此外進行反向MR分析,以探究食管癌和與食管癌存在因果關系的腸道菌群之間是否存在反向因果關系。
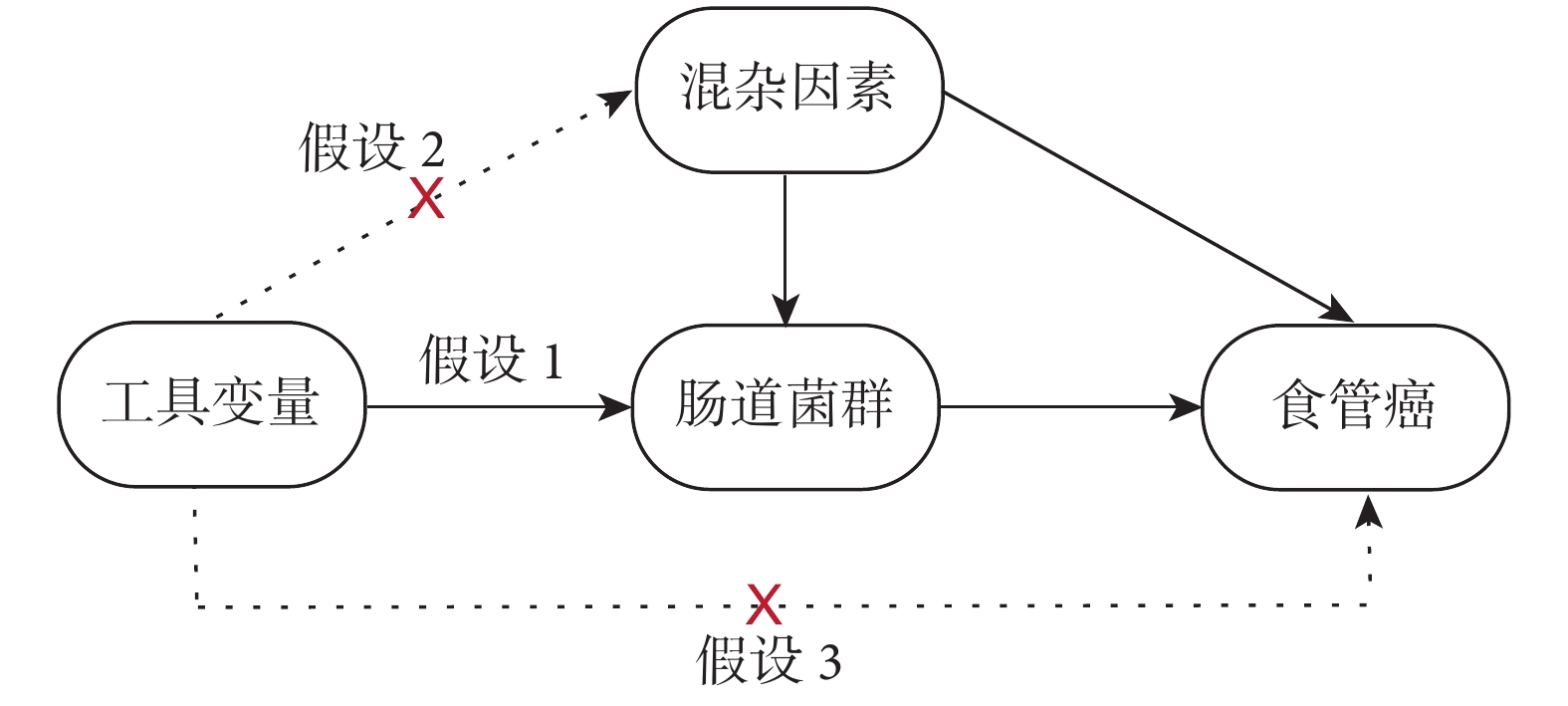
本研究需遵循MR研究方法的3種關鍵假設:(1)IVs與腸道菌群之間存在顯著關聯;(2)IVs與腸道菌群-食管癌的所有混雜因素均不相關;(3)IVs只能通過腸道菌群影響食管癌,而不是通過其他任何途徑;見圖1。
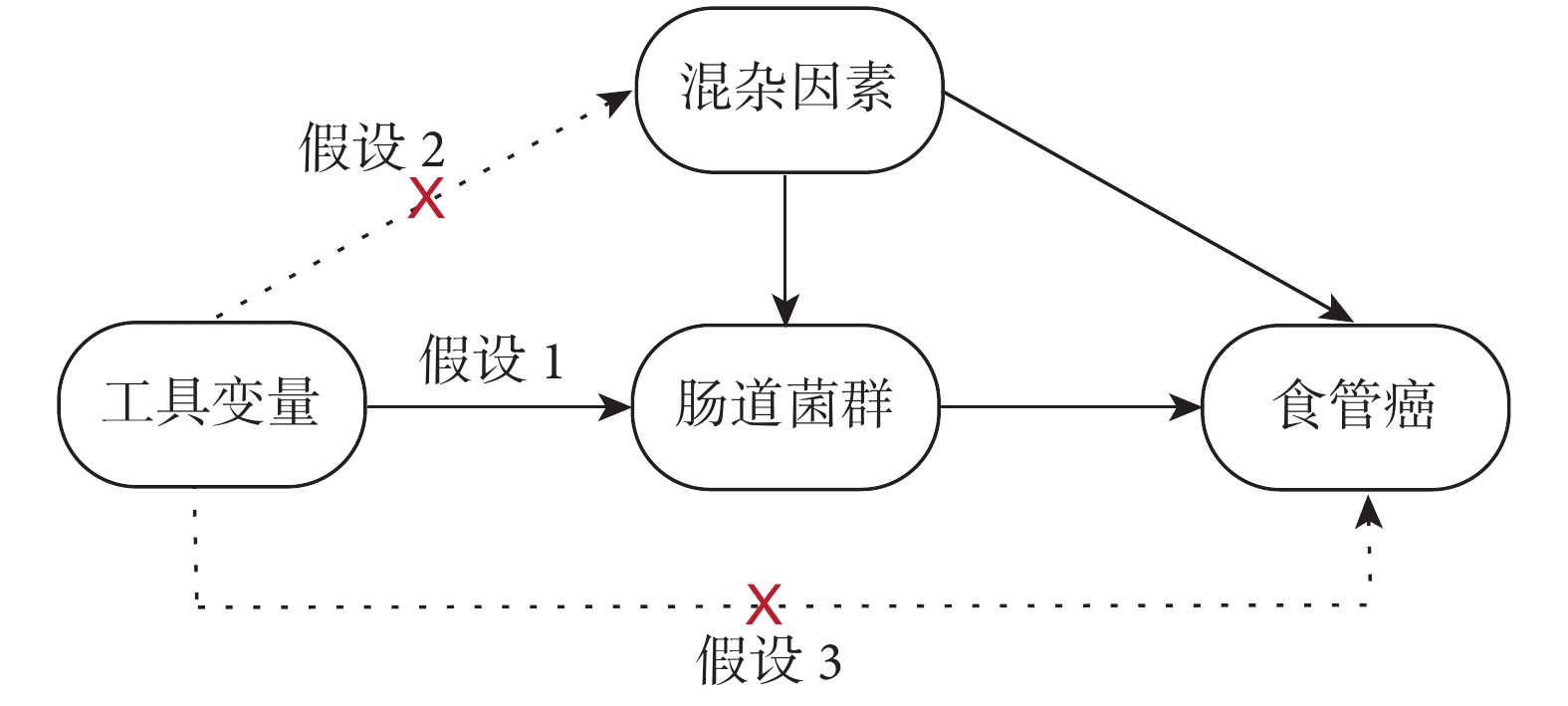 圖1
孟德爾隨機化分析的假設圖
圖1
孟德爾隨機化分析的假設圖
1.2 數據來源
腸道菌群遺傳數據來源于規模最大的MiBioGen聯盟發表的GWAS,MiBioGen聯盟收集了來自歐洲、美國等11個國家的18340例(歐洲血統13266例)受試者的16s RNA基因測序譜和基因分型數據,對腸道微生物組特征位點進行分析[22]。該分析共包括211個細菌分類群,屬是最低的細菌分類水平,包括15個未知屬,因此我們將196個細菌分類群納入本次研究[23-24]。食管癌遺傳數據來自GWAS摘要數據中的英國生物樣本數據(UK Biobank),包括740例食管癌患者和372016例對照患者,均為歐洲血統。由于使用的是公開數據庫,因此不再提供原始數據列表。
1.3 工具變量選擇
研究預先根據全位點顯著性閾值P<5.0×10?8選擇與腸道菌群相關的SNP,由于某些腸道菌群的SNP無法提取或個數<3個,因此我們放寬了閾值要求,根據P<1.0×10?5選擇顯著相關的SNP[25-26]。根據以下質量控制條件進一步篩選SNP,以確保腸道菌群與食管癌之間存在因果關系的準確性:(1)排除暴露和結局樣本之間等位基因不一致的SNP,如A/C;(2)去除回文SNP;(3)設置系數r2閾值0.001,設定區域寬度10000 kb,以排除連鎖不平衡的干擾;(4)去除等位基因頻率(minor allele frequency,MAF)<0.01的SNP;(5)通過phenoscanner網站(www.phenoscanner.medschl.cam.ac.uk)查詢并去除與暴露-結局有關的混雜因素的SNP,包括吸煙、飲酒、體質量指數等[5, 27],如 rs10167839(既往吸煙史),rs3734633(體質量指數)。MR-Egger回歸和孟德爾隨機多態性殘差和離群值(Mendelian randomization pleiotropy residual sum and outlier,MR-PRESSO)檢驗潛在的水平多效性[28-29]。MR-Egger回歸用于評估整體SNP的平均水平多效性,而MR-PRESSO可以檢測每個異常SNP,并通過去除異常值來消除水平多效性,即通過去除具有水平多效性的SNP重新進行MR分析。對于顯著相關的SNP,我們采用F統計量來評估所選擇的遺傳變量是否為弱IVs,F>10表示遺傳變量不存在弱IVs偏倚[30],F統計量計算公式為R2(n?k?1)/k(1?R2),其中n代表暴露樣本數量,k代表SNP的數量,R2代表SNP解釋的變異的占比。
1.4 孟德爾隨機化分析
本研究采用MR-隨機或固定效應逆方差加權法(inverse variance weighted,IVW)、加權中位數估計(weighted median estimation,WME)、MR-Egger回歸、單一模式(simple mode,SM)和加權模式(weighted mode,WM)5種方法驗證196個類群的腸道菌群與食管癌之間是否存在因果關系。IVW方法是使用Wald比值法進行單個SNP的關聯,然后再選擇固定效應或隨機效應模型對多個位點效應進行Meta匯總,能夠提供最準確的效應估計值[18]。對于僅包含1個SNP的IVs,采用Wald比值法進行MR分析[31]。WME方法的前提是基于至少50%的IVs是有效假設,給出準確的評估結果[31]。MR-Egger回歸考慮到截距項的存在,可以檢測和調整水平多效性,如果不存在水平多效性,那么MR-Egger回歸和IVW的結果基本一致[18]。SM和WM方法也是MR分析中的2種重要統計學方法[32]。本研究以IVW分析方法為主,其他方法作為補充。
1.5 敏感性分析
為進一步檢驗研究結果的準確性和穩定性,我們使用異質性檢驗、多效性水平檢驗和留一法(leave-one-out)進行敏感性分析。Cochrane’s Q 檢驗用于評估每個細菌相關的SNP的異質性,如果存在異質性(P≤0.05),則使用隨機IVW方法;如果不存在異質性(P>0.05),則使用固定IVW方法。留一法用于評估MR因果關系是否由單個SNP驅動。MR-Egger回歸檢驗的截距項與0差異很大時,說明存在水平多效性[18, 33],此時需要用MR-PRESSO剔除異常SNP來消除水平多效性,并重新進行MR分析。
1.6 統計學分析
本研究使用R軟件(版本4.2.0)的TwosampleMR包和MR-PRESSO R包進行數據分析,P≤0.05為差異有統計學意義。
2 結果
2.1 孟德爾隨機化結果
根據IVs的篩選標準,對196個類群的腸道微生物的GWAS數據進行SNP篩選,所有SNP的F統計量均>10,不存在弱IVs偏倚,最終用于MR分析的是8個類群的72個SNP(SNP特征見附件表1,https://www.tcsurg.org/article/10.7507/1007-4848.202403016)。
IVW結果表明,8個類群的腸道微生物與食管癌存在因果關系,其中草酸桿菌科(P=0.023)、糞桿菌屬(P=0.028)、塞內加爾菌屬(P=0.006)和韋榮氏球菌屬(P=0.018)可能是食管癌的危險因素,伯克霍爾德氏菌目(P=0.002)、真桿菌屬氧化還原類(P=0.038)、羅姆布茨菌屬(P=0.048)和蘇黎世桿菌屬(P=0.013)可能是食管癌的保護因素(表1)。圖2顯示了5種MR分析方法的研究結果,所有的Beta方向一致。
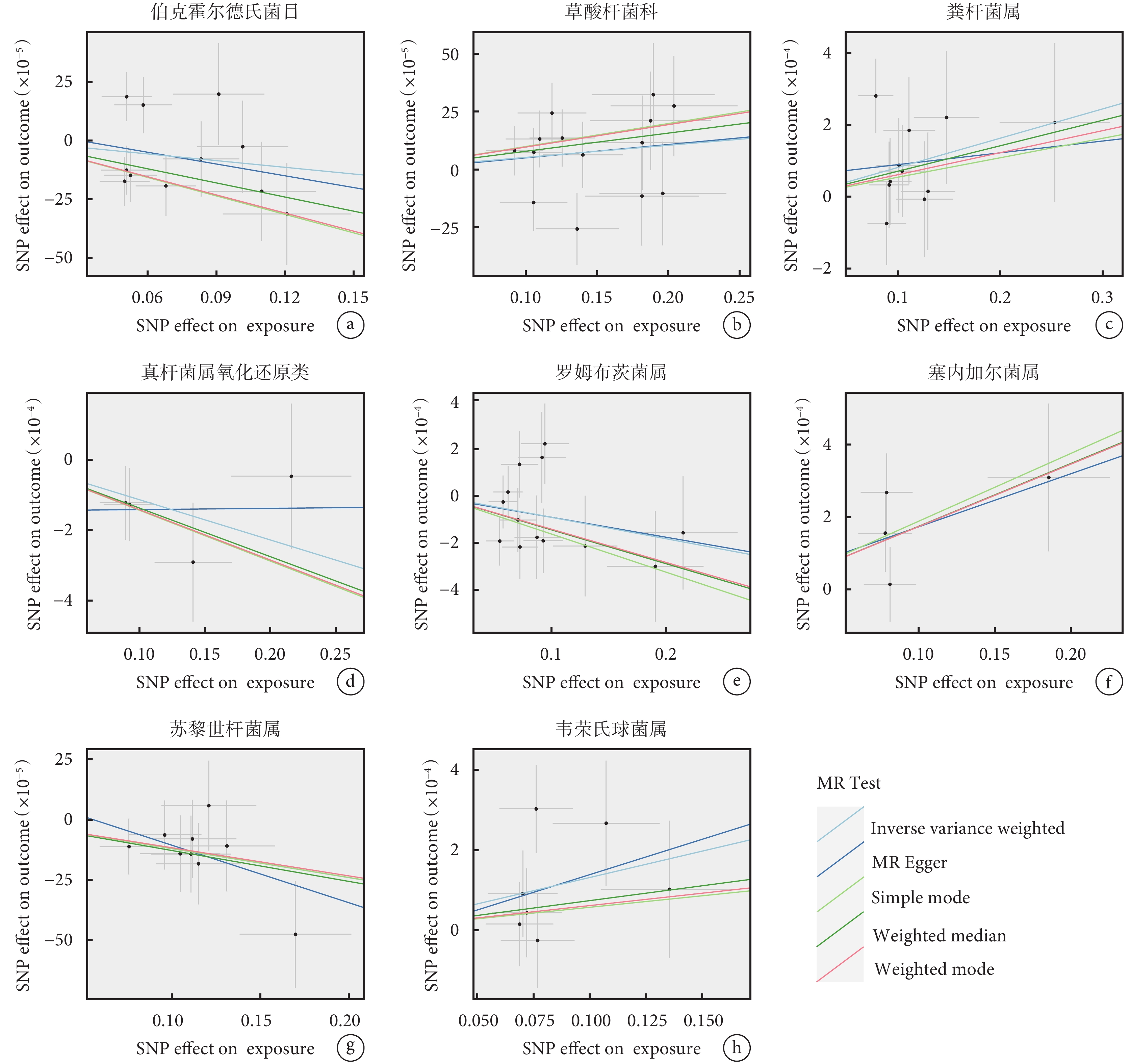 圖2
腸道菌群與食管癌孟德爾隨機化分析的散點圖
圖2
腸道菌群與食管癌孟德爾隨機化分析的散點圖
a~h:分別為伯克霍爾德氏菌目、草酸桿菌科、糞桿菌屬、真桿菌屬氧化還原類、羅姆布茨菌屬、塞內加爾菌屬、蘇黎世桿菌屬、韋榮氏球菌屬與食管癌之間相關性的散點圖;SNP:單核苷酸多態性
2.2 敏感性分析
對8個類群的腸道微生物進行Cochrane’s Q異質性檢驗,P均>0.05,即不存在異質性。MR-Egger回歸檢驗的截距項和0進行統計檢驗,P均>0.05,MR-PRESSO檢驗未發現異常值,P>0.05,表明不存在水平多效性(表2)。
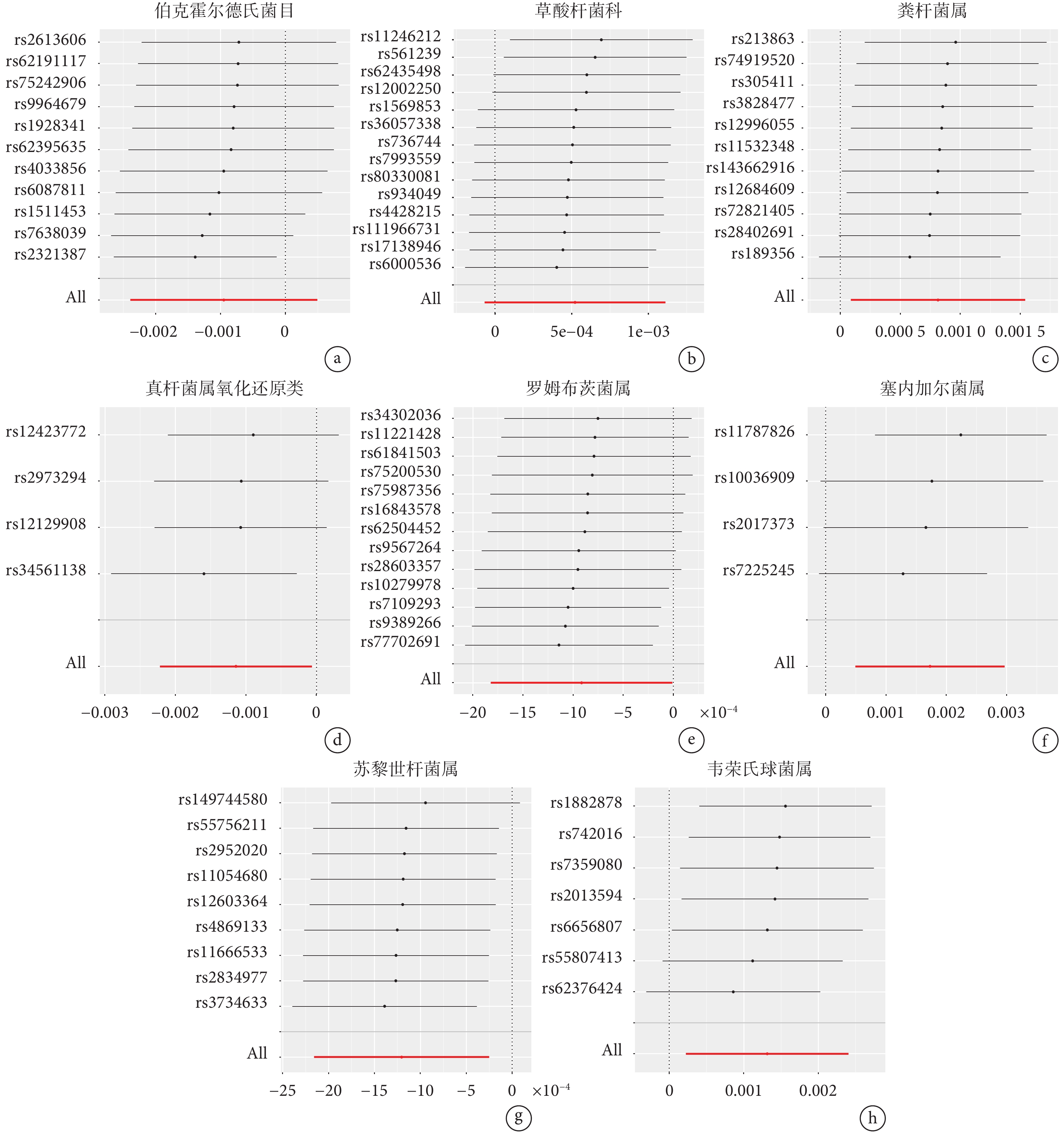
留一法分析發現,納入的IVs的效應值和總效應值大小較為接近,尚未發現對因果關系有顯著影響的SNP(圖3)。上述結果表明,8個類群的腸道微生物與食管癌之間的因果關系穩定。
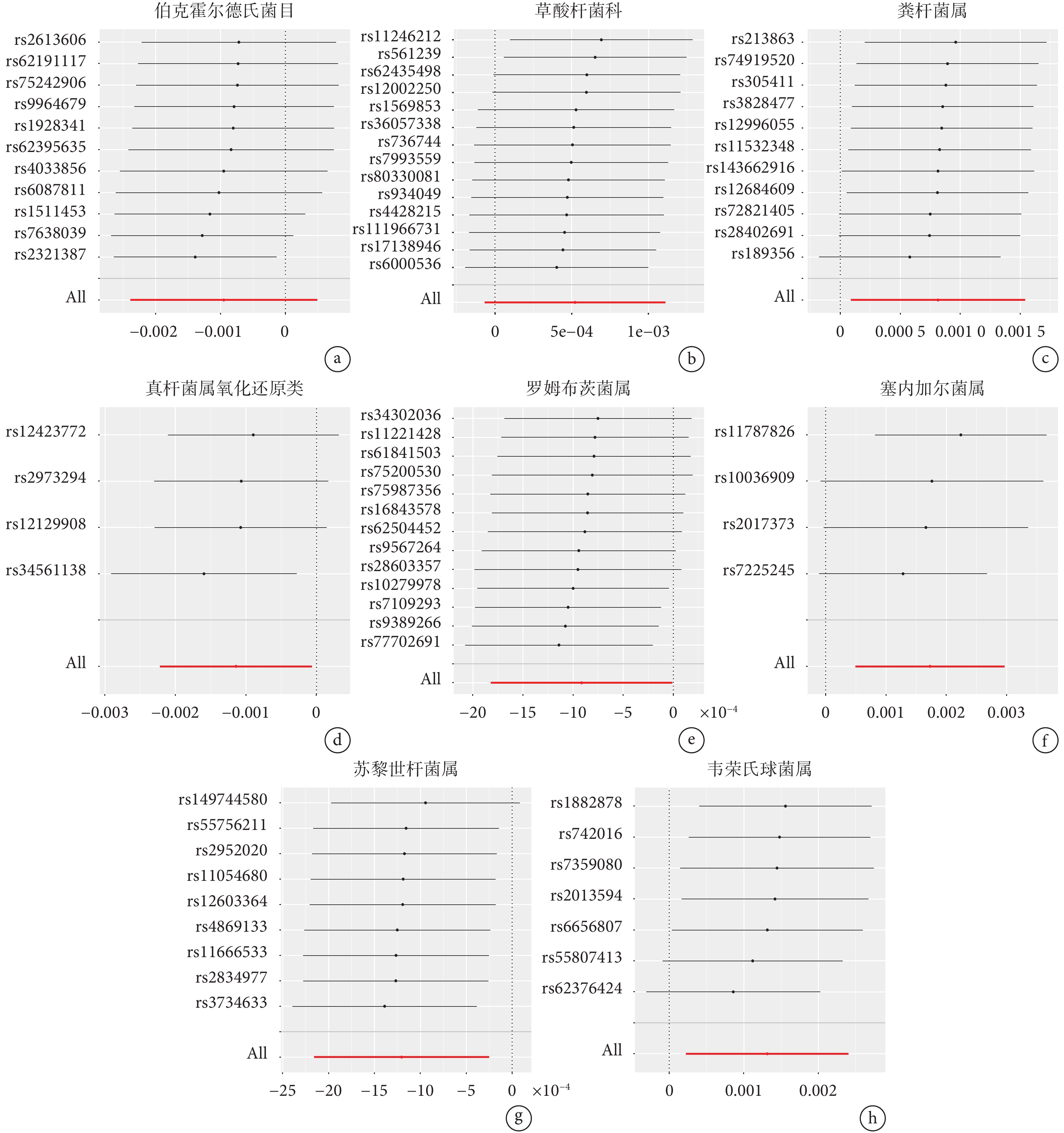 圖3
腸道菌群與食管癌的留一法敏感性分析結果
圖3
腸道菌群與食管癌的留一法敏感性分析結果
a~h:分別為伯克霍爾德氏菌目、草酸桿菌科、糞桿菌屬、真桿菌屬氧化還原類、羅姆布茨菌屬、塞內加爾菌屬、蘇黎世桿菌屬、韋榮氏球菌屬與食管癌之間相關性的留一法敏感性分析
2.3 反向孟德爾隨機化分析
以食管癌作為暴露,與食管癌有因果關系的腸道微生物(草酸桿菌科、糞桿菌屬、塞內加爾菌屬、韋榮氏球菌屬、伯克霍爾德氏菌目、真桿菌屬氧化還原類、羅姆布茨菌屬、蘇黎世桿菌屬)作為結局進行了反向MR分析,研究并未發現顯著的因果關系,表3僅展示IVW方法分析結果。敏感性分析未發現異質性和水平多效性的存在(表4)。
3 討論
本研究分析了腸道菌群與食管癌之間是否存在因果關系,結果表明,草酸桿菌科、糞桿菌屬、塞內加爾菌屬和韋榮氏球菌屬與食管癌發病風險增加有關,而伯克霍爾德氏菌目、真桿菌屬氧化還原類、羅姆布茨菌屬和蘇黎世桿菌屬則是食管癌的保護因素。
腸道微生物可能通過多種機制影響食管癌,這些機制涉及人體免疫系統和信號通路之間的復雜作用[34-36]。一方面,腸道微生物主要通過炎癥反應和富含生態失調的菌群產生致癌化合物來介導致癌效應[37-41],如通過炎癥反應產生的硝酸鹽可促進腸桿菌科等兼性厭氧菌富集,引起微生物失調-炎癥-微生物失調的惡性循環[42]。此外,糞腸球菌和糞梭桿菌分別會產生超氧化物和硫化氫,從而導致雙鏈 DNA 斷裂和染色體不穩定[42-43];彎曲桿菌屬和螺旋桿菌屬通過產生遺傳性毒性化合物(如細胞致死性擴張毒素和大腸桿菌素)來調節致癌作用[44-45]。雖然韋榮氏球菌屬在食管癌中的作用機制研究較少,但研究[36, 46]已明確指出,與健康對照組相比,食管癌患者的韋榮氏球菌屬豐度顯著增加,這與本研究結果一致。另一方面,腸道微生物可能會通過產生短鏈脂肪酸(如丁酸鹽)或調節抗腫瘤免疫力來介導腫瘤抑制作用[41]。真桿菌氧化還原類菌群通過合成丁酸鹽抑制腫瘤進展[47-48]。伯克霍爾德氏菌可作為一種益生菌來調節腸道微生物群的多樣性,從而提高抗腫瘤免疫力[49]。Deng 等[50]對23例食管癌患者進行的一項16s RNA 基因測序分析發現,食管癌患者的羅姆布茨菌屬豐度較健康對照組顯著增加,這與本研究結果[OR=0.999,95%CI(0.998,1.000),P=0.048]存在差異,有必要進行多中心對照試驗和基礎實驗研究來探索羅姆布茨菌屬的作用機制。
本研究首次表明蘇黎世桿菌對食管癌具有保護作用。越來越多的研究表明,蘇黎世桿菌與多種疾病有關,包括結腸癌[51]、食管上皮萎縮[52]、虹膜黃素病[52]、胃癌[53]、胰腺癌[54]和創傷后骨關節炎[55]。中草藥抗癌雞尾酒湯能引起蘇黎世桿菌豐度顯著提高,進而通過Th17途徑激活免疫細胞,導致結腸癌細胞的破壞[51]。蘇黎世桿菌與免疫調節細胞有關,或許這正是蘇黎世桿菌對食管癌具有保護作用的原因,但其確切作用仍需進一步探索。值得注意的是,本研究發現了2種未報道過的與食管癌相關的細菌,即塞內加爾菌屬和草酸桿菌科,它們都是食管癌的危險因素。目前研究僅發現塞內加爾菌屬與慢性腎臟病[26]和多囊卵巢綜合征[56]有關。草酸桿菌科與多種疾病相關,包括多囊卵巢綜合征[57]、結直腸癌[58]、膽道癌[59]、炎癥性腸病[60]和克羅恩病[60]等。雖然目前尚無文獻揭示這2種細菌與食管癌之間的關系,但本研究提示它們可能是食管癌進展的危險因素,值得進一步探索研究。
本研究的主要優點是減少了混雜因素和反向因果關系對結果的干擾,這比觀察性研究更具有說服力,提高了研究結果的可信度。然而,這項研究也存在一些局限性。首先,由于使用的數據是在線匯總統計數據,因此無法對包括性別在內的差異進行亞組分析,這可能會帶來潛在偏倚。其次,為獲得SNP,我們將遺傳變異的閾值設定為 P<1.0×10?5,可能會造成弱IVs偏倚。第三,雖然GWAS中的腸道菌群數據大多是歐洲血統,但必須考慮到人群分層干擾的可能性,從而導致估計結果偏倚和影響普遍性。
綜上所述,本研究采用了雙樣本雙向MR分析來探討腸道菌群和食管癌的因果關系。研究結果表明二者存在因果關系。雖然腸道菌群在食管癌發病過程中的作用機制尚不清楚,但本研究為未來腸道菌群與食管癌之間的研究提供了參考。
利益沖突:無。
作者貢獻:汪夢夢、高銘駿負責選題與研究設計,撰寫論文;周嗣丁、田澍雨負責統計分析,論文修改;束余聲、王霄霖負責文章審閱和定稿。
食管癌(esophageal cancer,EC)是第8大常見癌癥,也是全球癌癥死亡的第6大原因,嚴重影響全球人類健康[1]。食管癌的5年生存率很低,約為12%~20%[2]。吸煙和飲酒是食管癌的兩大危險因素[3-6]。此外,遺傳易感性、體質量指數增加、必需微量營養素攝入、口腔感染和飲食改變等因素也是食管癌高發的誘因[7-10]。目前,食管癌的治療采用包括手術、化學治療、放射治療和免疫靶向治療在內的綜合治療為主[11],但針對食管癌病因的干預方式較少,及時針對病因進行干預降低食管癌發生率,有助于改善食管癌患者的生存。
腸道菌群與人體新陳代謝、免疫調節和神經內分泌系統密切相關[12]。健康人90% 以上的細菌來自厚壁菌門和擬桿菌門,其次是疣微菌門、變形菌門和放線菌門,共占微生物的99%[13]。越來越多的證據表明,食管癌患者的特定細菌和細菌菌群失調,可通過破壞DNA、激活致癌信號通路、產生促瘤代謝物和抑制抗腫瘤免疫來促進食管癌進展[14]。Zhou等[15]發現,高豐度的產酸細菌(主要是乳酸桿菌、雙歧桿菌、葡萄球菌和鏈球菌)可通過乳酸代謝失調促進致癌作用。Münch等[16]使用Barrett食管的動物模型發現,高脂肪飲食誘導的腸道微生物群變化導致促炎細胞因子和免疫細胞水平升高,繼而導致促腫瘤免疫表型。腸道微生物可起保護作用,其擾動和水平降低能促進炎性反應和縮短生存時間[17]。根據這些研究,塑造腸道微生物群的組成可能有望實現有效和特異性的抗腫瘤免疫反應。基于此,我們從孟德爾遺傳角度出發,對腸道菌群和食管癌的關系進行研究。
與觀察性研究不同,孟德爾隨機化(Mendelian randomization,MR)分析是使用遺傳變異作為工具變量(instrumentalvariables,IVs)來推斷暴露與結果之間的因果關系,遺傳變異和結局之間不受混雜因素的影響,可作為研究腸道微生物與食管癌之間因果關系的一種新方法[18]。MR已被廣泛用于研究腸道微生物與疾病之間的關聯,包括癌癥[19]、免疫性疾病[20]、代謝性疾病[21]。在該研究中,我們使用全基因組關聯研究(genome-wide association studies,GWAS)的匯總統計數據進行雙向MR分析,以評估腸道菌群和食管癌的因果關系。
1 資料與方法
1.1 研究設計
本研究將腸道菌群作為暴露因素,選取顯著相關的單核苷酸多態性(single-nucleotide polymorphism,SNP)作為IVs,食管癌作為結局變量。基于公開的GWAS匯總統計數據,使用雙樣本雙向MR分析方法評估196個細菌分類群與食管癌之間的因果關系。使用異質性檢驗和水平多效性檢驗等分析方法驗證結果的可靠性和穩定性。此外進行反向MR分析,以探究食管癌和與食管癌存在因果關系的腸道菌群之間是否存在反向因果關系。
本研究需遵循MR研究方法的3種關鍵假設:(1)IVs與腸道菌群之間存在顯著關聯;(2)IVs與腸道菌群-食管癌的所有混雜因素均不相關;(3)IVs只能通過腸道菌群影響食管癌,而不是通過其他任何途徑;見圖1。
圖1
孟德爾隨機化分析的假設圖
1.2 數據來源
腸道菌群遺傳數據來源于規模最大的MiBioGen聯盟發表的GWAS,MiBioGen聯盟收集了來自歐洲、美國等11個國家的18340例(歐洲血統13266例)受試者的16s RNA基因測序譜和基因分型數據,對腸道微生物組特征位點進行分析[22]。該分析共包括211個細菌分類群,屬是最低的細菌分類水平,包括15個未知屬,因此我們將196個細菌分類群納入本次研究[23-24]。食管癌遺傳數據來自GWAS摘要數據中的英國生物樣本數據(UK Biobank),包括740例食管癌患者和372016例對照患者,均為歐洲血統。由于使用的是公開數據庫,因此不再提供原始數據列表。
1.3 工具變量選擇
研究預先根據全位點顯著性閾值P<5.0×10?8選擇與腸道菌群相關的SNP,由于某些腸道菌群的SNP無法提取或個數<3個,因此我們放寬了閾值要求,根據P<1.0×10?5選擇顯著相關的SNP[25-26]。根據以下質量控制條件進一步篩選SNP,以確保腸道菌群與食管癌之間存在因果關系的準確性:(1)排除暴露和結局樣本之間等位基因不一致的SNP,如A/C;(2)去除回文SNP;(3)設置系數r2閾值0.001,設定區域寬度10000 kb,以排除連鎖不平衡的干擾;(4)去除等位基因頻率(minor allele frequency,MAF)<0.01的SNP;(5)通過phenoscanner網站(www.phenoscanner.medschl.cam.ac.uk)查詢并去除與暴露-結局有關的混雜因素的SNP,包括吸煙、飲酒、體質量指數等[5, 27],如 rs10167839(既往吸煙史),rs3734633(體質量指數)。MR-Egger回歸和孟德爾隨機多態性殘差和離群值(Mendelian randomization pleiotropy residual sum and outlier,MR-PRESSO)檢驗潛在的水平多效性[28-29]。MR-Egger回歸用于評估整體SNP的平均水平多效性,而MR-PRESSO可以檢測每個異常SNP,并通過去除異常值來消除水平多效性,即通過去除具有水平多效性的SNP重新進行MR分析。對于顯著相關的SNP,我們采用F統計量來評估所選擇的遺傳變量是否為弱IVs,F>10表示遺傳變量不存在弱IVs偏倚[30],F統計量計算公式為R2(n?k?1)/k(1?R2),其中n代表暴露樣本數量,k代表SNP的數量,R2代表SNP解釋的變異的占比。
1.4 孟德爾隨機化分析
本研究采用MR-隨機或固定效應逆方差加權法(inverse variance weighted,IVW)、加權中位數估計(weighted median estimation,WME)、MR-Egger回歸、單一模式(simple mode,SM)和加權模式(weighted mode,WM)5種方法驗證196個類群的腸道菌群與食管癌之間是否存在因果關系。IVW方法是使用Wald比值法進行單個SNP的關聯,然后再選擇固定效應或隨機效應模型對多個位點效應進行Meta匯總,能夠提供最準確的效應估計值[18]。對于僅包含1個SNP的IVs,采用Wald比值法進行MR分析[31]。WME方法的前提是基于至少50%的IVs是有效假設,給出準確的評估結果[31]。MR-Egger回歸考慮到截距項的存在,可以檢測和調整水平多效性,如果不存在水平多效性,那么MR-Egger回歸和IVW的結果基本一致[18]。SM和WM方法也是MR分析中的2種重要統計學方法[32]。本研究以IVW分析方法為主,其他方法作為補充。
1.5 敏感性分析
為進一步檢驗研究結果的準確性和穩定性,我們使用異質性檢驗、多效性水平檢驗和留一法(leave-one-out)進行敏感性分析。Cochrane’s Q 檢驗用于評估每個細菌相關的SNP的異質性,如果存在異質性(P≤0.05),則使用隨機IVW方法;如果不存在異質性(P>0.05),則使用固定IVW方法。留一法用于評估MR因果關系是否由單個SNP驅動。MR-Egger回歸檢驗的截距項與0差異很大時,說明存在水平多效性[18, 33],此時需要用MR-PRESSO剔除異常SNP來消除水平多效性,并重新進行MR分析。
1.6 統計學分析
本研究使用R軟件(版本4.2.0)的TwosampleMR包和MR-PRESSO R包進行數據分析,P≤0.05為差異有統計學意義。
2 結果
2.1 孟德爾隨機化結果
根據IVs的篩選標準,對196個類群的腸道微生物的GWAS數據進行SNP篩選,所有SNP的F統計量均>10,不存在弱IVs偏倚,最終用于MR分析的是8個類群的72個SNP(SNP特征見附件表1,https://www.tcsurg.org/article/10.7507/1007-4848.202403016)。
IVW結果表明,8個類群的腸道微生物與食管癌存在因果關系,其中草酸桿菌科(P=0.023)、糞桿菌屬(P=0.028)、塞內加爾菌屬(P=0.006)和韋榮氏球菌屬(P=0.018)可能是食管癌的危險因素,伯克霍爾德氏菌目(P=0.002)、真桿菌屬氧化還原類(P=0.038)、羅姆布茨菌屬(P=0.048)和蘇黎世桿菌屬(P=0.013)可能是食管癌的保護因素(表1)。圖2顯示了5種MR分析方法的研究結果,所有的Beta方向一致。
圖2
腸道菌群與食管癌孟德爾隨機化分析的散點圖
a~h:分別為伯克霍爾德氏菌目、草酸桿菌科、糞桿菌屬、真桿菌屬氧化還原類、羅姆布茨菌屬、塞內加爾菌屬、蘇黎世桿菌屬、韋榮氏球菌屬與食管癌之間相關性的散點圖;SNP:單核苷酸多態性
2.2 敏感性分析
對8個類群的腸道微生物進行Cochrane’s Q異質性檢驗,P均>0.05,即不存在異質性。MR-Egger回歸檢驗的截距項和0進行統計檢驗,P均>0.05,MR-PRESSO檢驗未發現異常值,P>0.05,表明不存在水平多效性(表2)。
留一法分析發現,納入的IVs的效應值和總效應值大小較為接近,尚未發現對因果關系有顯著影響的SNP(圖3)。上述結果表明,8個類群的腸道微生物與食管癌之間的因果關系穩定。
圖3
腸道菌群與食管癌的留一法敏感性分析結果
a~h:分別為伯克霍爾德氏菌目、草酸桿菌科、糞桿菌屬、真桿菌屬氧化還原類、羅姆布茨菌屬、塞內加爾菌屬、蘇黎世桿菌屬、韋榮氏球菌屬與食管癌之間相關性的留一法敏感性分析
2.3 反向孟德爾隨機化分析
以食管癌作為暴露,與食管癌有因果關系的腸道微生物(草酸桿菌科、糞桿菌屬、塞內加爾菌屬、韋榮氏球菌屬、伯克霍爾德氏菌目、真桿菌屬氧化還原類、羅姆布茨菌屬、蘇黎世桿菌屬)作為結局進行了反向MR分析,研究并未發現顯著的因果關系,表3僅展示IVW方法分析結果。敏感性分析未發現異質性和水平多效性的存在(表4)。
3 討論
本研究分析了腸道菌群與食管癌之間是否存在因果關系,結果表明,草酸桿菌科、糞桿菌屬、塞內加爾菌屬和韋榮氏球菌屬與食管癌發病風險增加有關,而伯克霍爾德氏菌目、真桿菌屬氧化還原類、羅姆布茨菌屬和蘇黎世桿菌屬則是食管癌的保護因素。
腸道微生物可能通過多種機制影響食管癌,這些機制涉及人體免疫系統和信號通路之間的復雜作用[34-36]。一方面,腸道微生物主要通過炎癥反應和富含生態失調的菌群產生致癌化合物來介導致癌效應[37-41],如通過炎癥反應產生的硝酸鹽可促進腸桿菌科等兼性厭氧菌富集,引起微生物失調-炎癥-微生物失調的惡性循環[42]。此外,糞腸球菌和糞梭桿菌分別會產生超氧化物和硫化氫,從而導致雙鏈 DNA 斷裂和染色體不穩定[42-43];彎曲桿菌屬和螺旋桿菌屬通過產生遺傳性毒性化合物(如細胞致死性擴張毒素和大腸桿菌素)來調節致癌作用[44-45]。雖然韋榮氏球菌屬在食管癌中的作用機制研究較少,但研究[36, 46]已明確指出,與健康對照組相比,食管癌患者的韋榮氏球菌屬豐度顯著增加,這與本研究結果一致。另一方面,腸道微生物可能會通過產生短鏈脂肪酸(如丁酸鹽)或調節抗腫瘤免疫力來介導腫瘤抑制作用[41]。真桿菌氧化還原類菌群通過合成丁酸鹽抑制腫瘤進展[47-48]。伯克霍爾德氏菌可作為一種益生菌來調節腸道微生物群的多樣性,從而提高抗腫瘤免疫力[49]。Deng 等[50]對23例食管癌患者進行的一項16s RNA 基因測序分析發現,食管癌患者的羅姆布茨菌屬豐度較健康對照組顯著增加,這與本研究結果[OR=0.999,95%CI(0.998,1.000),P=0.048]存在差異,有必要進行多中心對照試驗和基礎實驗研究來探索羅姆布茨菌屬的作用機制。
本研究首次表明蘇黎世桿菌對食管癌具有保護作用。越來越多的研究表明,蘇黎世桿菌與多種疾病有關,包括結腸癌[51]、食管上皮萎縮[52]、虹膜黃素病[52]、胃癌[53]、胰腺癌[54]和創傷后骨關節炎[55]。中草藥抗癌雞尾酒湯能引起蘇黎世桿菌豐度顯著提高,進而通過Th17途徑激活免疫細胞,導致結腸癌細胞的破壞[51]。蘇黎世桿菌與免疫調節細胞有關,或許這正是蘇黎世桿菌對食管癌具有保護作用的原因,但其確切作用仍需進一步探索。值得注意的是,本研究發現了2種未報道過的與食管癌相關的細菌,即塞內加爾菌屬和草酸桿菌科,它們都是食管癌的危險因素。目前研究僅發現塞內加爾菌屬與慢性腎臟病[26]和多囊卵巢綜合征[56]有關。草酸桿菌科與多種疾病相關,包括多囊卵巢綜合征[57]、結直腸癌[58]、膽道癌[59]、炎癥性腸病[60]和克羅恩病[60]等。雖然目前尚無文獻揭示這2種細菌與食管癌之間的關系,但本研究提示它們可能是食管癌進展的危險因素,值得進一步探索研究。
本研究的主要優點是減少了混雜因素和反向因果關系對結果的干擾,這比觀察性研究更具有說服力,提高了研究結果的可信度。然而,這項研究也存在一些局限性。首先,由于使用的數據是在線匯總統計數據,因此無法對包括性別在內的差異進行亞組分析,這可能會帶來潛在偏倚。其次,為獲得SNP,我們將遺傳變異的閾值設定為 P<1.0×10?5,可能會造成弱IVs偏倚。第三,雖然GWAS中的腸道菌群數據大多是歐洲血統,但必須考慮到人群分層干擾的可能性,從而導致估計結果偏倚和影響普遍性。
綜上所述,本研究采用了雙樣本雙向MR分析來探討腸道菌群和食管癌的因果關系。研究結果表明二者存在因果關系。雖然腸道菌群在食管癌發病過程中的作用機制尚不清楚,但本研究為未來腸道菌群與食管癌之間的研究提供了參考。
利益沖突:無。
作者貢獻:汪夢夢、高銘駿負責選題與研究設計,撰寫論文;周嗣丁、田澍雨負責統計分析,論文修改;束余聲、王霄霖負責文章審閱和定稿。