

肺癌是21世紀全球發生率和死亡率最高的惡性腫瘤,每年約有209萬新發病例和176萬死亡病例,嚴重威脅著人類生命健康[1-2]。其中,肺腺癌(lung adenocarcinoma,LUAD)是最普遍的組織病理學類型,發病率逐年上升[3]。目前,手術切除仍是治療LUAD最有效的方法。雖然隨著治療方法的多元化發展,晚期肺癌患者的生存狀況顯著改善,然而,其5年總生存率依然低于20%[4]。腫瘤微環境是由腫瘤、基質和內皮細胞組成的異質、復雜的組織,其特點是腫瘤與先天性和適應性免疫細胞之間的交互作用[5]。同時,研究[6]發現免疫微環境可通過促進腫瘤血管生成或調節腫瘤干細胞活性等方式直接或間接影響腫瘤的發生、發展。近年來,免疫治療作為一種新興治療模式,在LUAD治療中的潛力較大。目前針對免疫檢查點PD1/PD-L1、CTLA4的抗體已經廣泛應用于LUAD的治療[7]。同時,隨著免疫治療的進展,已有多項臨床試驗CheckMate-032 (NCT01928394)[8]、KEYNOTE-010(NCT01905657)[9]證實了Nivolumab、Pembrolizumab、Atezolizumab 較化療均顯著延長患者總生存期。然而,LUAD的免疫治療仍存在許多困境,如耐藥性增多、不良反應頻發、特異性低、療效出現瓶頸等問題,阻礙了其進一步發展。因此,挖掘安全有效的免疫相關的潛在作用靶點對于改善LUAD免疫治療困境來說具有重要意義。
近年來,利用生物信息學分析腫瘤免疫微環境,挖掘腫瘤生物標志物是一種流行而有效的方法。例如,利用WGCNA對乳腺癌進展和預后的研究[10]表明,有一個基因模塊與患者生存時間相關,并且該研究通過共表達網絡分析確定了8個候選生物標志物,為進一步了解乳腺癌的分子發病機制提供了依據。也有研究[11]通過ssGSEA將The Cancer Genome Atlas(TCGA)數據庫中舌鱗狀細胞癌患者分類為不同免疫細胞侵蝕的集群,并利用ESTIMATE和CIBERSORT分析對集群之間的免疫狀況進行評估,最終確定了15個免疫相關基因可以作為舌鱗狀細胞癌患者預后的潛在生物標志物。由此可見,通過生物信息學的分析方法挖掘癌癥治療的潛在生物標志物已受到眾多研究者的關注。
本研究通過WGCNA和ESTIMATE等生物信息學分析方法對TCGA數據庫來源的LUAD表達數據進行分析,最終獲得與免疫細胞相關的hub基因。同時,基于hub基因構建了預后風險模型,并評估了RiskScore對免疫治療益處的預測能力。總的來說,本研究的結果有利于尋找LUAD潛在的診斷或治療生物標志物,這對于臨床為患者提供個性化的治療策略具有重要意義。
1 資料與方法
1.1 數據來源
LUAD mRNA表達數據集(FPKM)TCGA-LUAD和對應的臨床信息從TCGA數據庫(
1.2 肺腺癌樣本免疫微環境分析
利用R包“estimate”[12]對TCGA-LUAD數據集中LUAD腫瘤樣本腫瘤微環境進行評估。
1.3 加權基因共表達網絡分析
首先通過R包“WGCNA”來建立基因共表達網絡[13]。然后通過使用 Pearson 相關函數測量 TCGA-LUAD 數據中基因之間的共表達。通過將共表達相似度提高到β=5來構建鄰接矩陣。從鄰接矩陣出發,構建拓撲重疊矩陣(TOM)來考慮拓撲相似性,并建立相應的不相似矩陣(1-TOM)來形成聚類。使用hclust函數利用不同矩陣進行分層聚類。最后基于拓撲重疊矩陣進行層次聚類,按照動態混合切割方法,每個模塊最少基因數目設置為50,相異性閾值設置為0.25。計算由第一主成分(PC1)表示的模塊特征基因表達式(ME),確定每個模塊的表達式。臨床資料與模塊特征基因表達相關,并確定基因顯著性(gene significance,GS)。通過將模塊的ME與模塊的基因表達相關聯,確定模塊成員的定量方法,顏色被隨機分配給模塊,最終選擇與免疫評分、基質評分和ESTIMATE評分顯著相關的模塊作為后續的研究對象。為了進一步探究特征模塊內基因的生物學功能,利用R包“clusterProfiler”[14]對特征模塊內基因進行富集分析,特征模塊基因最顯著富集terms的篩選標準為P<0.05和Q<0.05。
1.4 獲取hub基因
本研究以cor.geneModuleMembership>0.8 和cor.geneTraitSignificance>0.6 為閾值篩選模塊內候選關鍵基因。同時,利用特征模塊內的所有基因在STRING (http://string-db.org/cgi/input.pl)網站構建 蛋白質-蛋白質相互作用(PPI)網絡,以相互作用評分>0.4 作為閾值,將獲得的候選hub基因與PPI 網絡中degree前30的基因取交集,最終獲得hub 基因。
1.5 hub基因的進一步驗證
首先比較hub基因在正常組中的表達差異。然后通過Pearson相關性分析hub 基因與各Stage分期之間的關系。Cancer Cell Line Encyclopedia(CCLE)數據庫(
1.6 評估RiskScore對免疫治療益處的預測能力
為了進一步分析RiskScore在預測治療益處中的作用。我們在獲得LUAD患者IPS數據的基礎上,將患者按風險評分的中位值分為高風險組和低風險組,利用t檢驗比較anti-PD1和anti-CTLA4 的治療效果在高低風險組中的差異(P<0.05),并利用R包“ggpubr”(
2 結果
2.1 WGCNA和特征模塊的識別
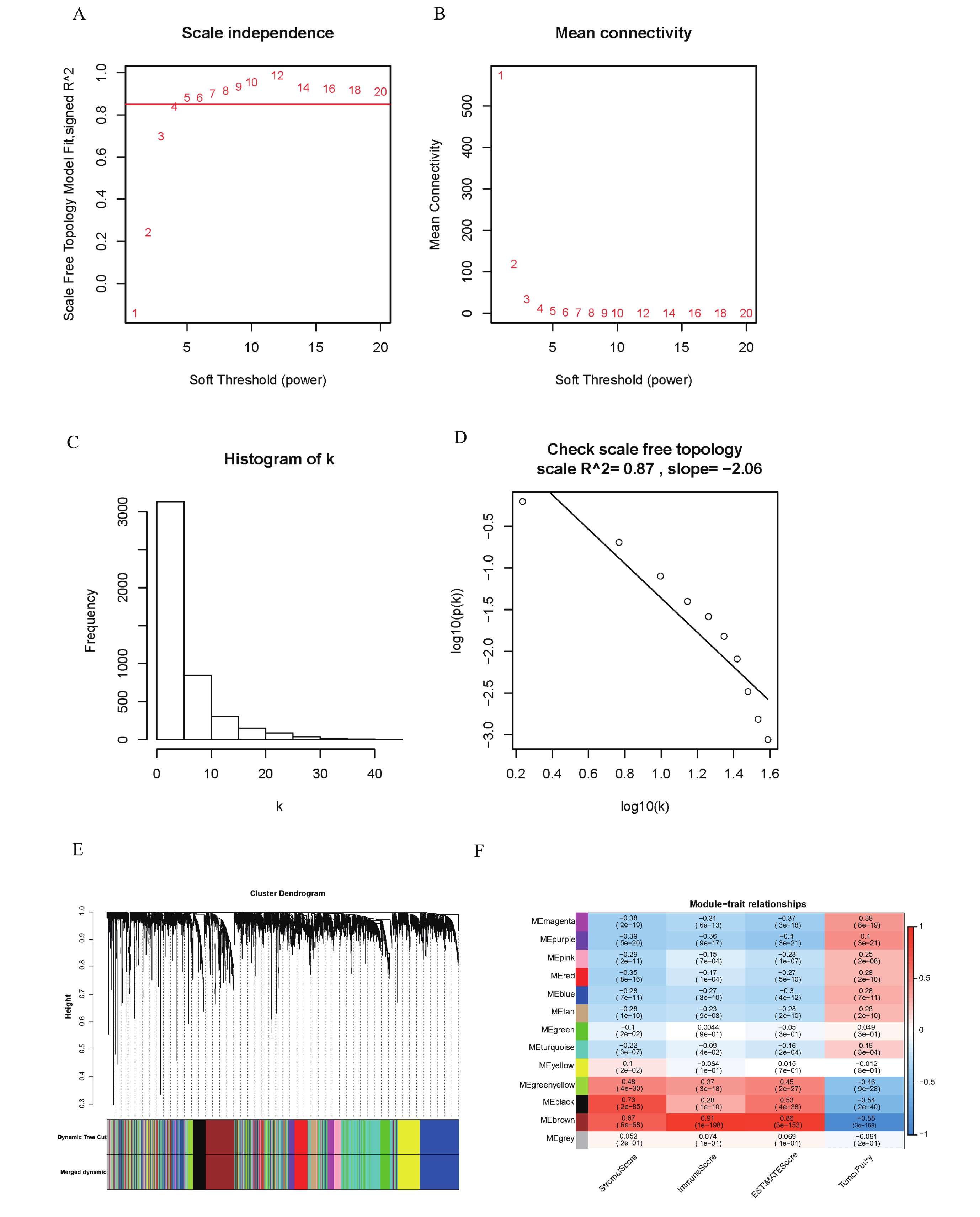
利用R包“WGCNA”對預處理后的511例腫瘤樣本MAD值最大的前5 000個基因構建加權基因共表達網絡。本研究中是以軟閾值為β=5來建立無尺度化網絡(圖1a~d)。接下來,基于平均層次聚類和動態樹切割,最終共獲得13個基因模塊(圖1e,表1)。同時,13個基因模塊和臨床性狀的相關性分析結果表明,brown模塊與StromalScore(r=0.67,P=6e-68)、ImmuneScore(r=0.91,P=1e-198)、ESTIMATEScore(r=0.86,P =3e-153)呈顯著正相關,與TumorPurity呈負相關(r=–0.88,P=3e-169)(圖1f),因此我們假設用于構建brown模塊的基因可能與免疫力有關。我們選擇brown模塊作為后續分析的研究對象。
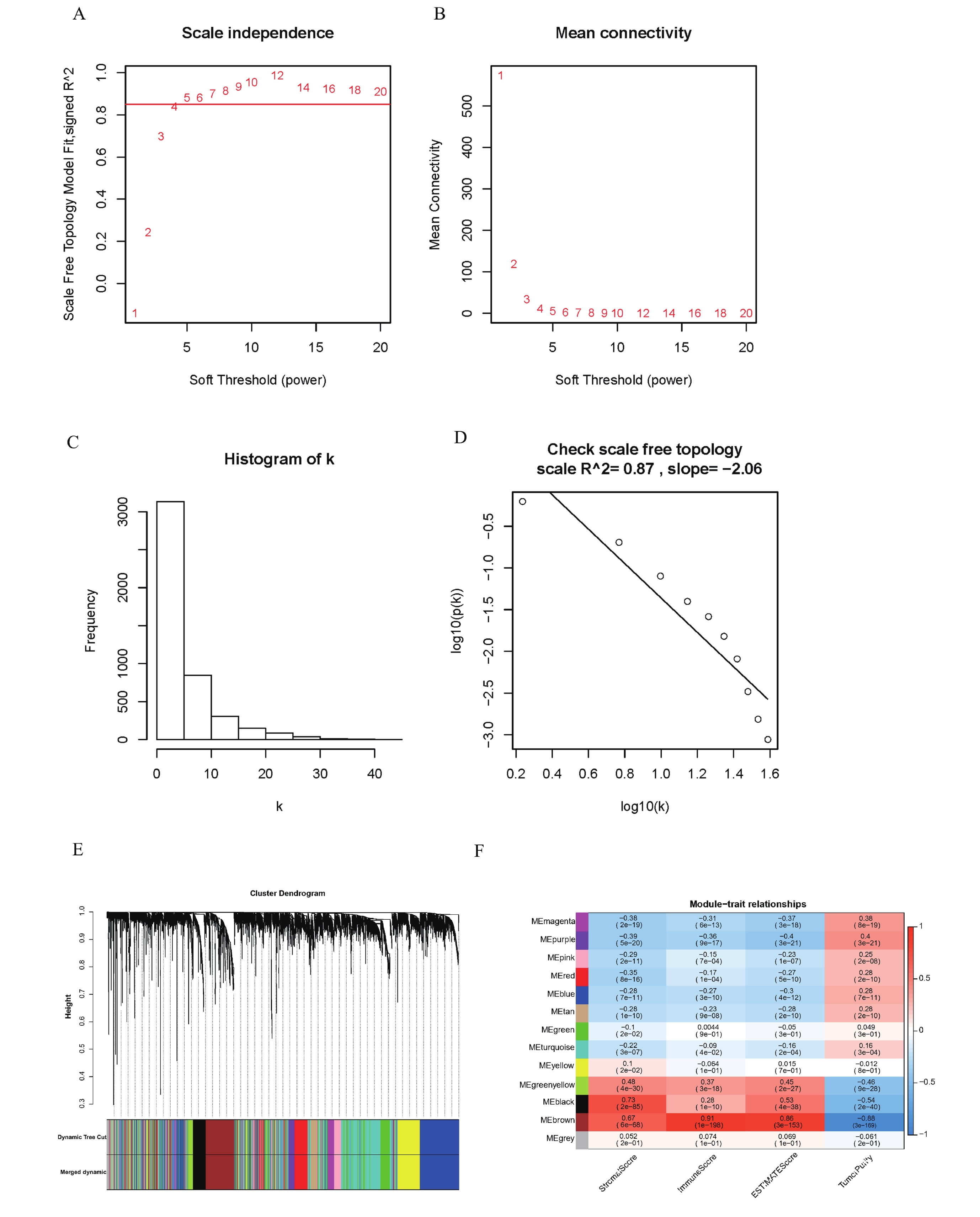 圖1
肺腺癌的加權基因共表達網絡
圖1
肺腺癌的加權基因共表達網絡
a:不同軟閾值功率下的無標度擬合指數分析(β);X軸表示軟閾值,Y軸表示不同軟閾值下的無標度擬合指數;紅線是對應于WGCNA分析的無標度擬合指數,最優軟閾值為5;b:不同軟閾值功率下的平均連通性;X軸表示軟閾值功率,Y軸反映平均連通性,表示不同軟閾值下的網絡連接性;c:β=5時連通性分布直方圖;d:檢查β=5時的無標度拓撲;e:基于不同度量(1-TOM)聚類的所有差異表達基因的樹狀圖,共13個不同的模塊,不同的顏色作為標識符標識不同的模塊;f:熱圖表示模塊hub基因與臨床性狀之間的相關性。ME表示模塊本征基因,它是在對模塊中的所有基因進行PCA分析后獲得的主成分1(PC1)的值;紅色表示與表型呈正相關,藍色表示與表型負相關
2.2 特征模塊內基因的生物功能和通路富集分析
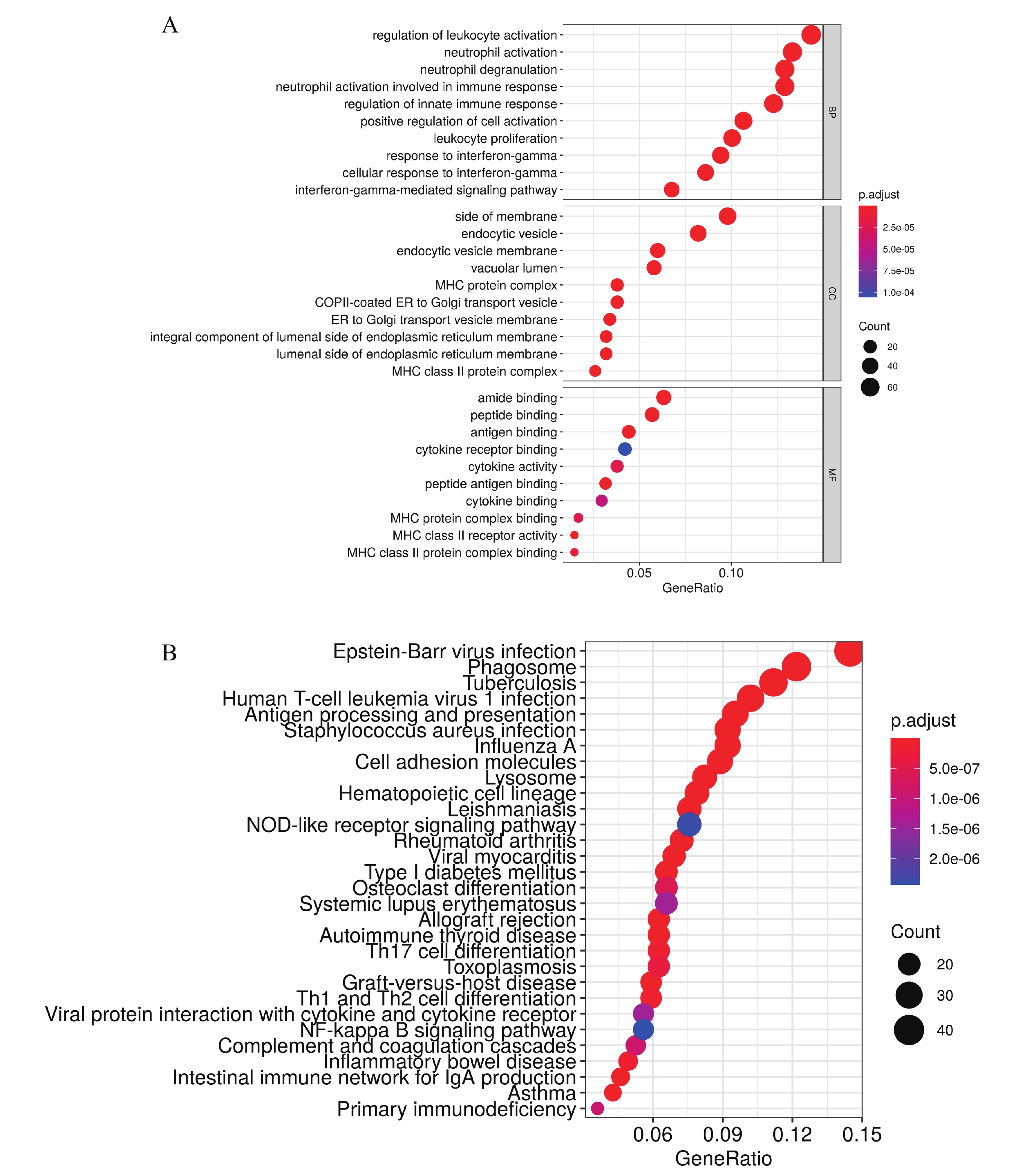
對brown模塊內的521個基因進行富集分析,結果表明該模塊內基因大多與中性粒細胞活化參與免疫反應、先天免疫反應的調節、細胞激活的正向調節、中性粒細胞活化和白細胞增殖等免疫功能相關;與lumenal side of endoplasmic reticulum membrane、integral component of lumenal side of endoplasmic reticulum membrane等內質網細胞組分相關;與MHC class Ⅱ protein complex binding和MHC class Ⅱ receptor activity等MHC受體活性有關(圖2a)。KEGG富集分析結果顯示該模塊內基因主要富集到NF-kappa B signaling pathway、NOD-like receptor signaling pathway、primary immunodeficiency、complement and coagulation cascade和human T-cell leukemia virus 1 infection等免疫和癌癥相關通路中(圖2b)。上述結果表明brown模塊中的基因主要與免疫功能相關,并在免疫相關途徑中富集。
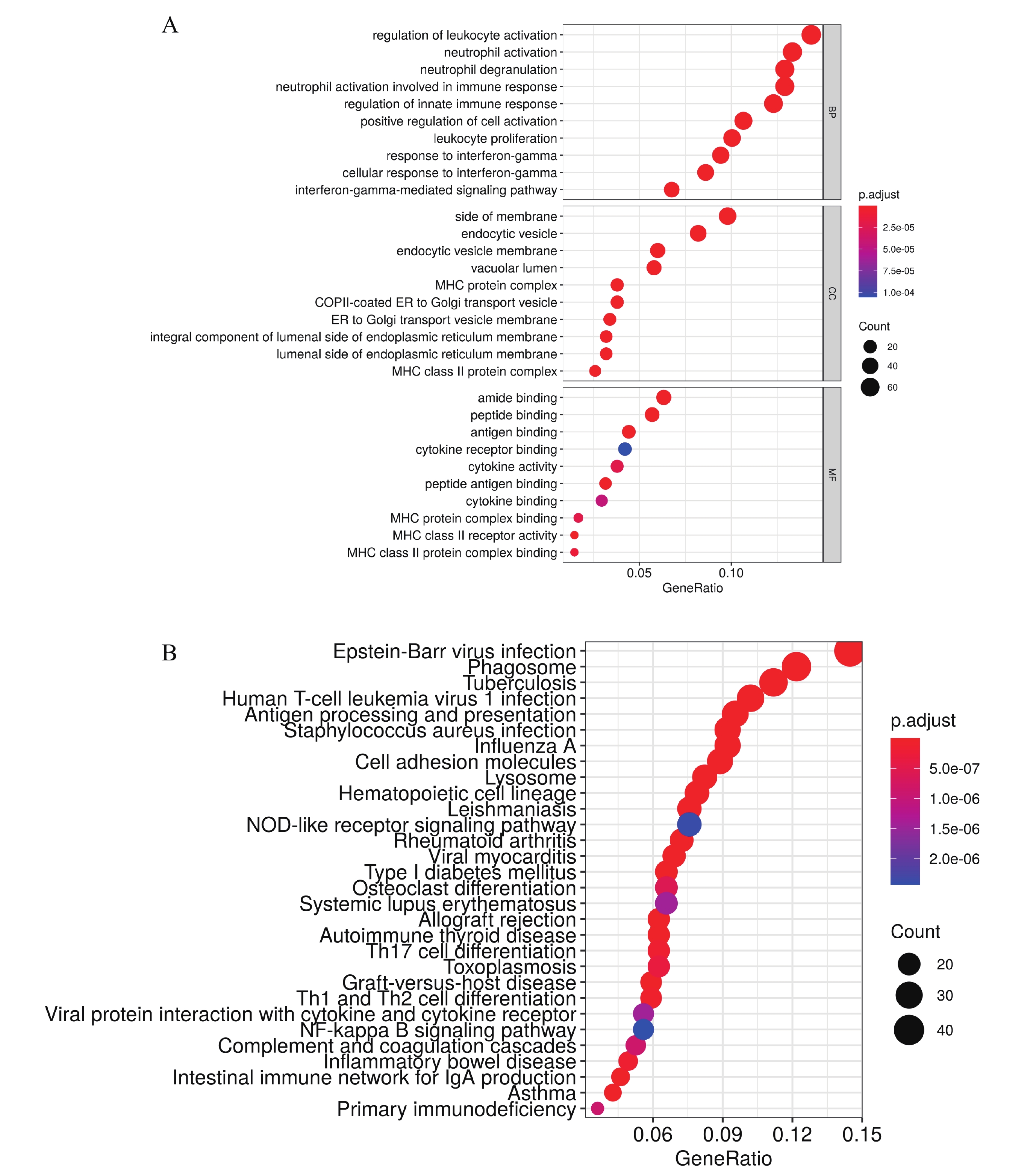 圖2
Brown模塊基因的富集分析
圖2
Brown模塊基因的富集分析
a:brown模塊中hub基因的GO功能富集分析,縱坐標表示不同的GO項,橫坐標表示基因富集數;b:brown模塊特征基因KEGG通路富集分析,縱坐標為KEGG通路注釋,橫坐標為基因富集數。 圖中的點的大小表示富集基因的數量;
2.3 與免疫相關的hub基因的鑒定
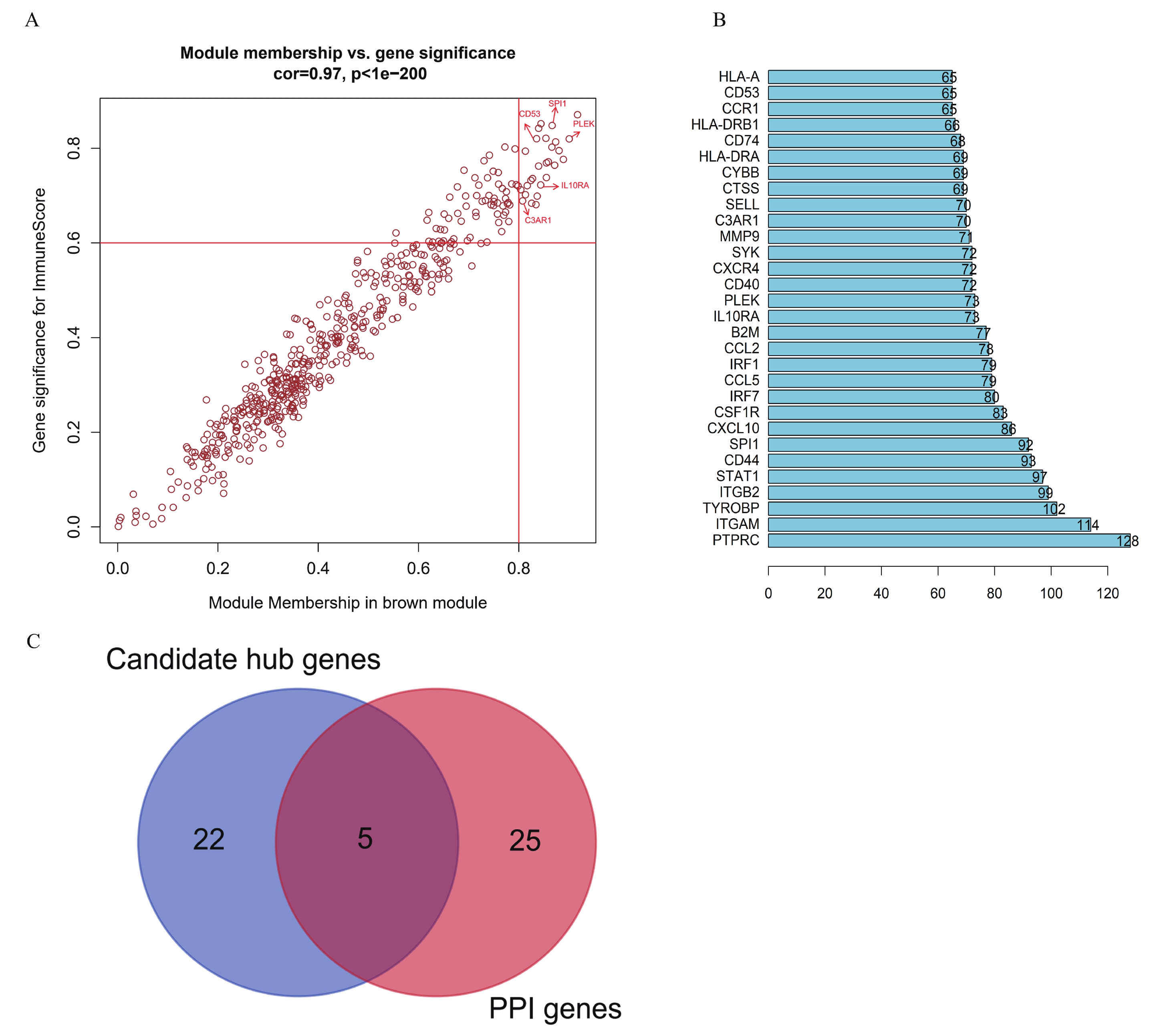
在brown基因模塊中篩選出與ImmuneScore具有較高相關性的27個基因作為候選hub 基因(圖3a)。同時,將brown模塊中的所有基因輸入STRING網站并構建PPI網絡,根據degree值篩選出前30基因(圖3b)。以相互作用評分>0.4作為閾值,將27個候選基因與前30基因取交集,最終獲得5個hub 基因,分別為CD53、PLEK、SPI1、IL10RA和C3AR1(圖3c)。
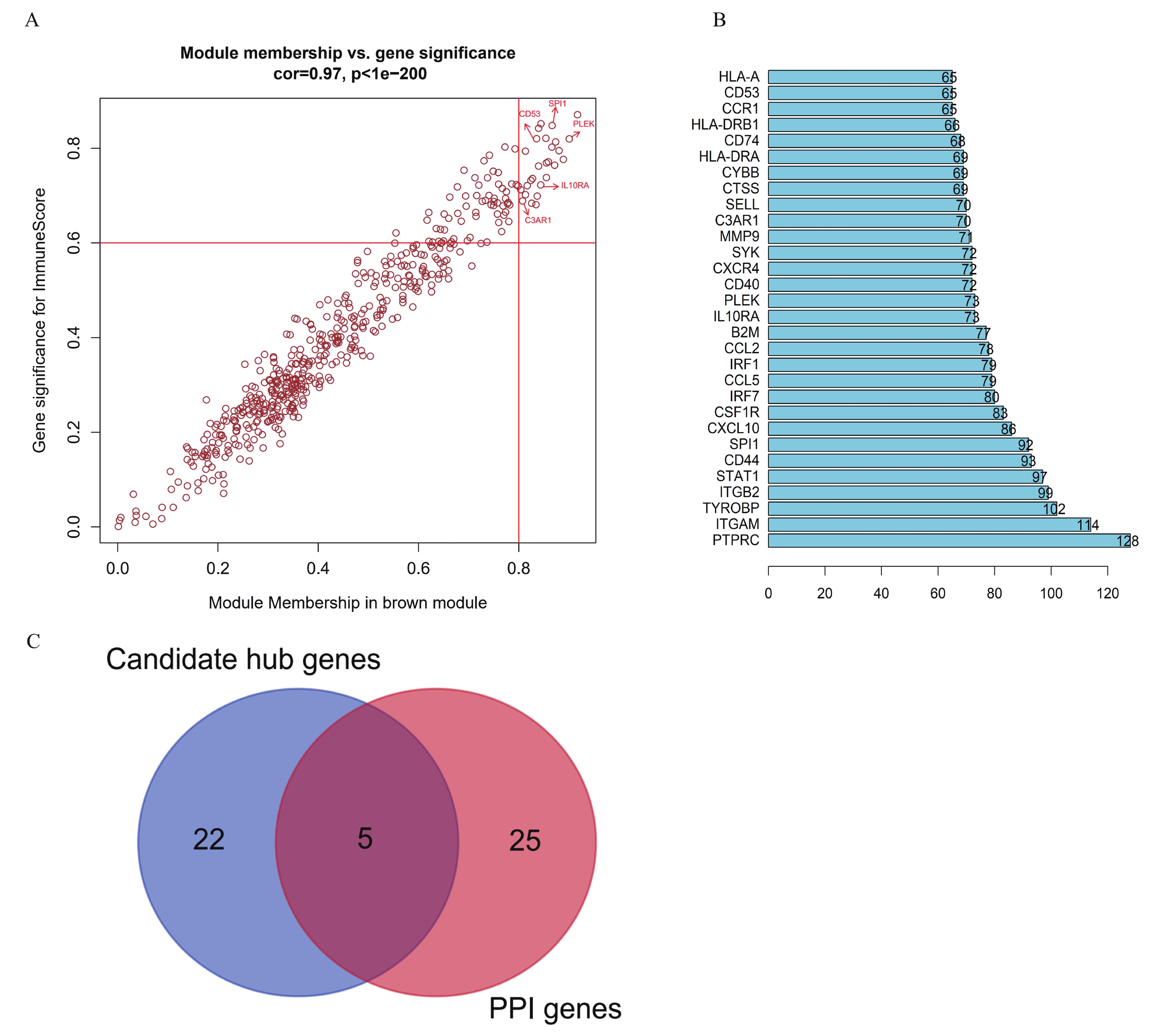 圖3
與免疫相關的hub基因的鑒定
圖3
與免疫相關的hub基因的鑒定
a:散點圖表示brown模塊中與組織學免疫評分相關的hub基因,X軸值表示基因和模塊之間的相關性;如果絕對值接近0,則該基因不是該模塊的一部分;如果這個值的絕對值接近1,那么該基因與該模塊高度相關。Y軸值表示基因和免疫評分之間的相關性;0表示該基因與免疫力不相關,1表示該基因與免疫力高度相關,圖中每個點代表一個基因;b:brown模塊中PPI網絡節點的前30個基因;c:韋恩圖篩選最終與免疫相關的關鍵基因;27個候選hub基因與PPI中degree前30個基因取交集
2.4 正常組與腫瘤組hub基因的表達差異及其與腫瘤分期的相關性及hub基因的Kaplan-Meier生存分析
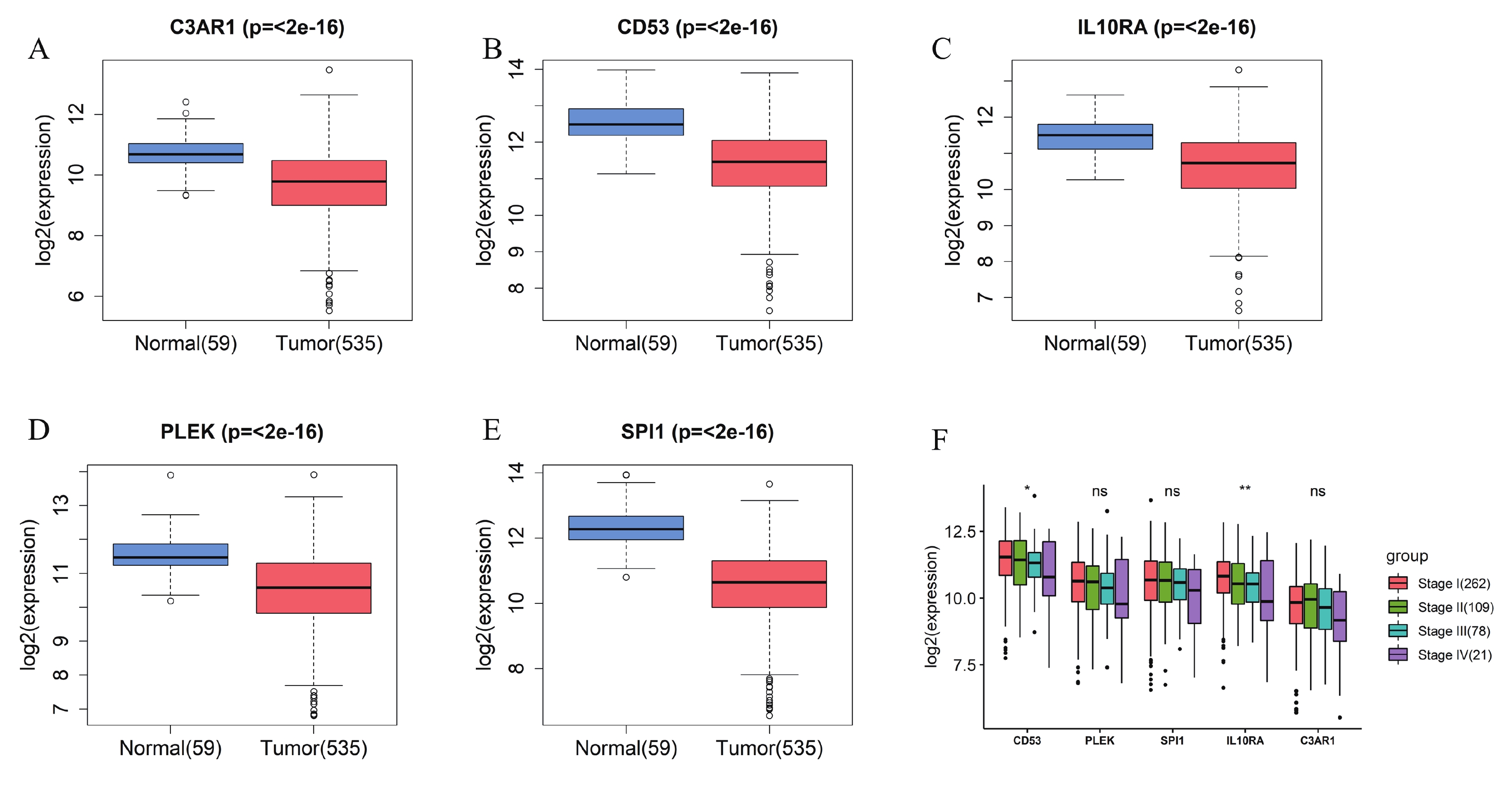
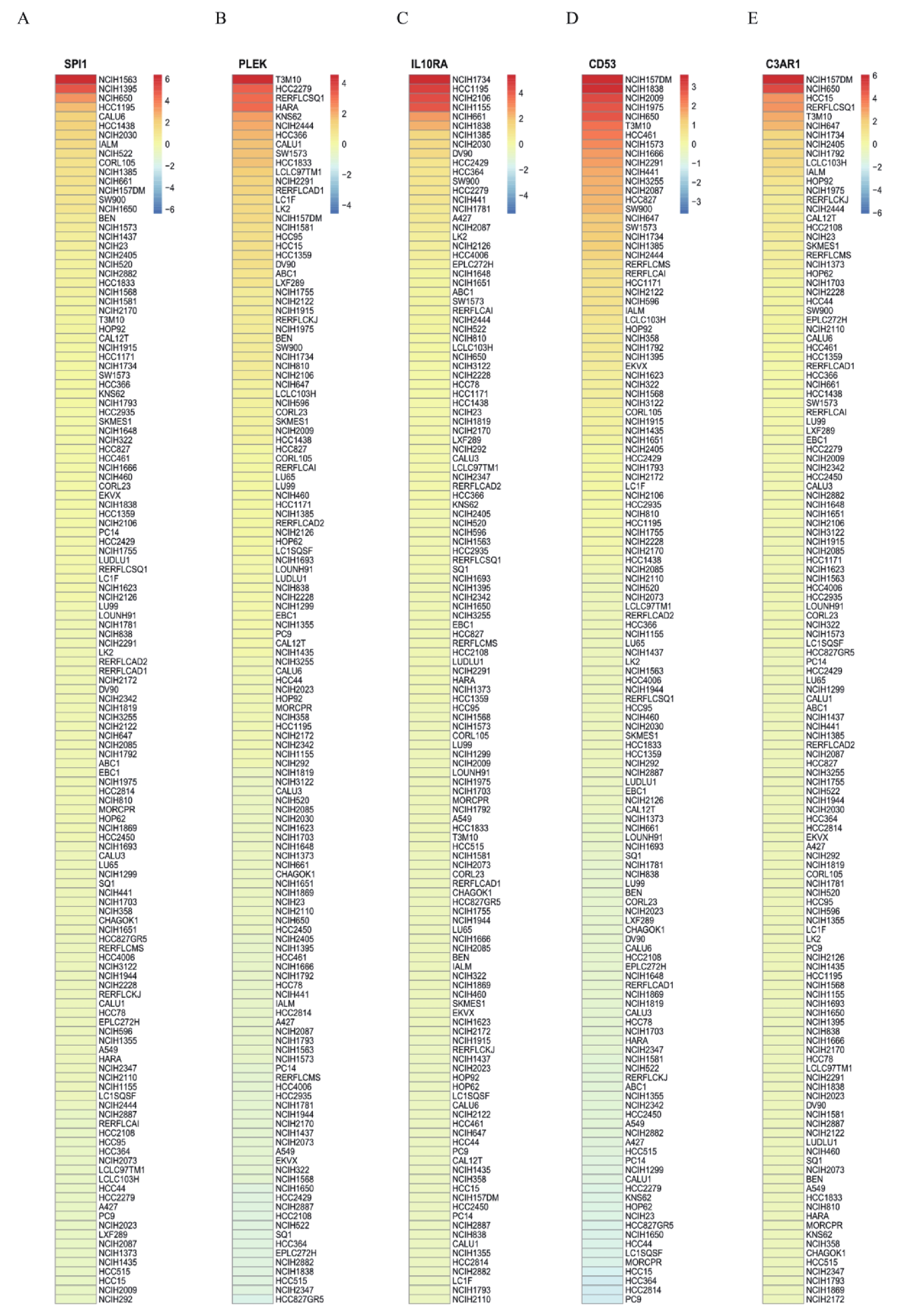
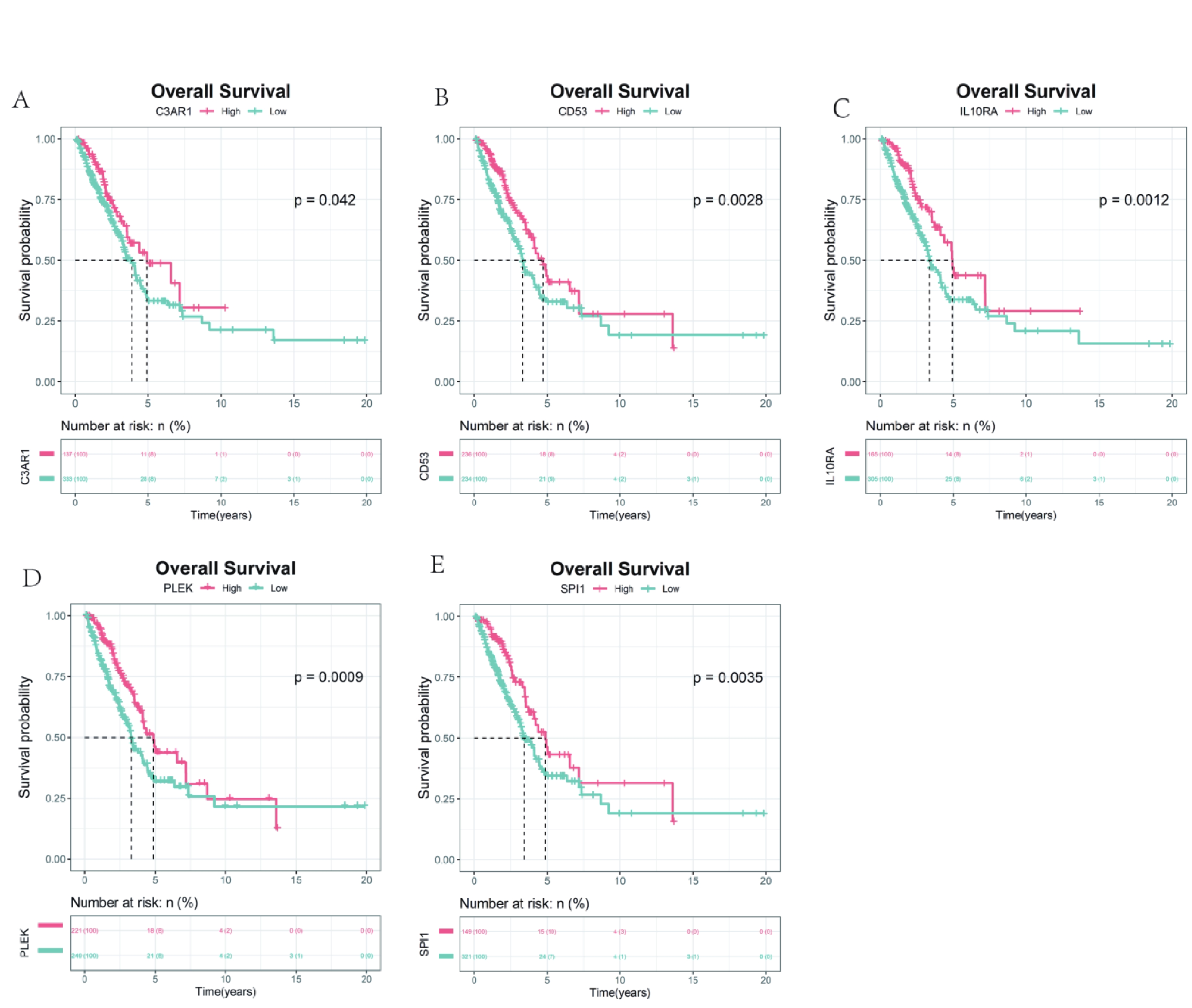
首先利用t檢驗分析了5個hub基因在正常組和腫瘤組中的表達差異,結果顯示,與正常組相比,5個hub 基因在腫瘤組中顯著低表達(圖4a~e)。同時,分析了5個hub基因的表達與Stage分期之間的相關性,結果顯示, CD53和IL10RA的表達水平與Stage分期有關聯(圖4f)。CCLE數據庫有大量的人類癌癥細胞系和獨特的數據集,能夠對大量人類癌癥細胞系進行詳細的遺傳表征[18]。因此,為了探索關鍵基因在大量LUAD細胞系中的表達差異,我們利用CCLE數據庫發現SPI1、PLEK、IL10RA、CD53和C3AR1在LUAD細胞株中的表達存在差異(圖5)。同時對患者的高低表達組進行生存分析,結果發現5個hub 基因的高表達預示著患者較好的預后(圖6)。
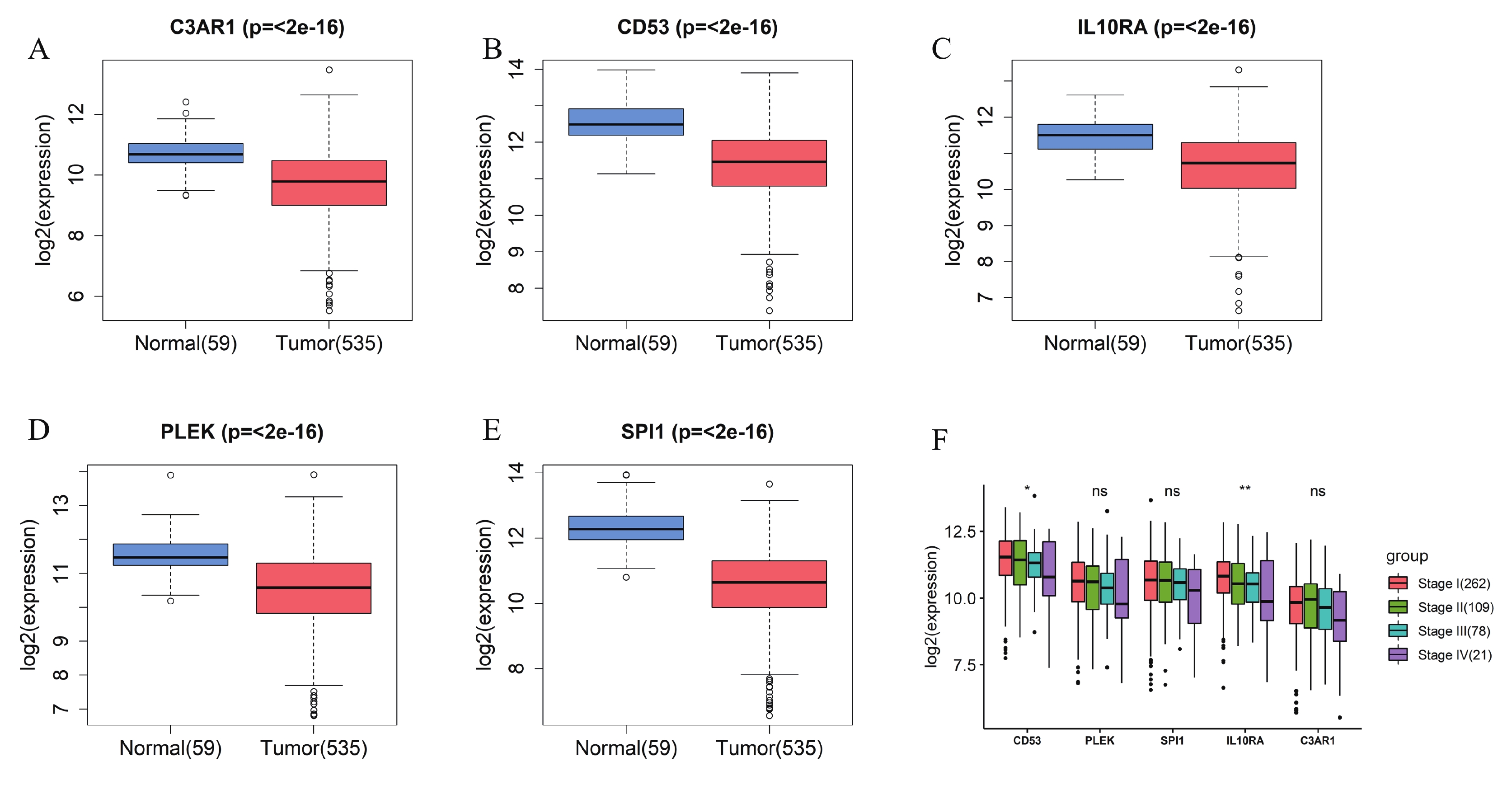 圖4
分析5個關鍵基因在正常組和腫瘤組的表達差異及在不同腫瘤分期中的mRNA表達變化
圖4
分析5個關鍵基因在正常組和腫瘤組的表達差異及在不同腫瘤分期中的mRNA表達變化
a~e:正常組和腫瘤組中C3AR1、CD53、IL10RA、PLEK和SPI1表達的差異;f:箱線圖表示5個hub基因在不同腫瘤分期中的mRNA表達情況。
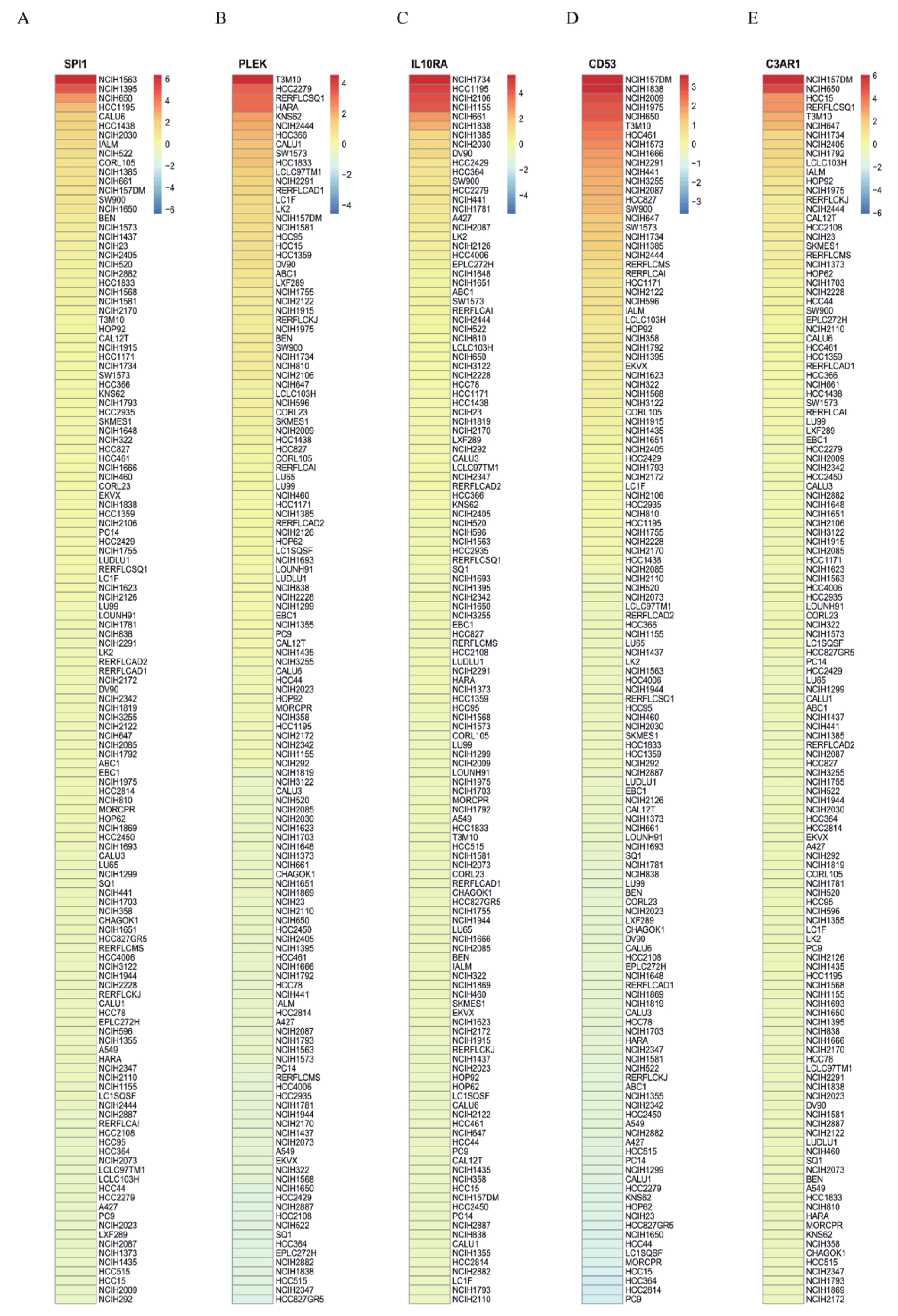 圖5
5個hub基因在大量肺腺癌細胞系中的表達
圖5
5個hub基因在大量肺腺癌細胞系中的表達
通過CCLE分析肺腺癌細胞系中SPI1(a)、PLEK(b)、IL10RA(c)、CD53(d)和C3AR1(e)的表達水平;紅色表示高表達,藍色表示低表達
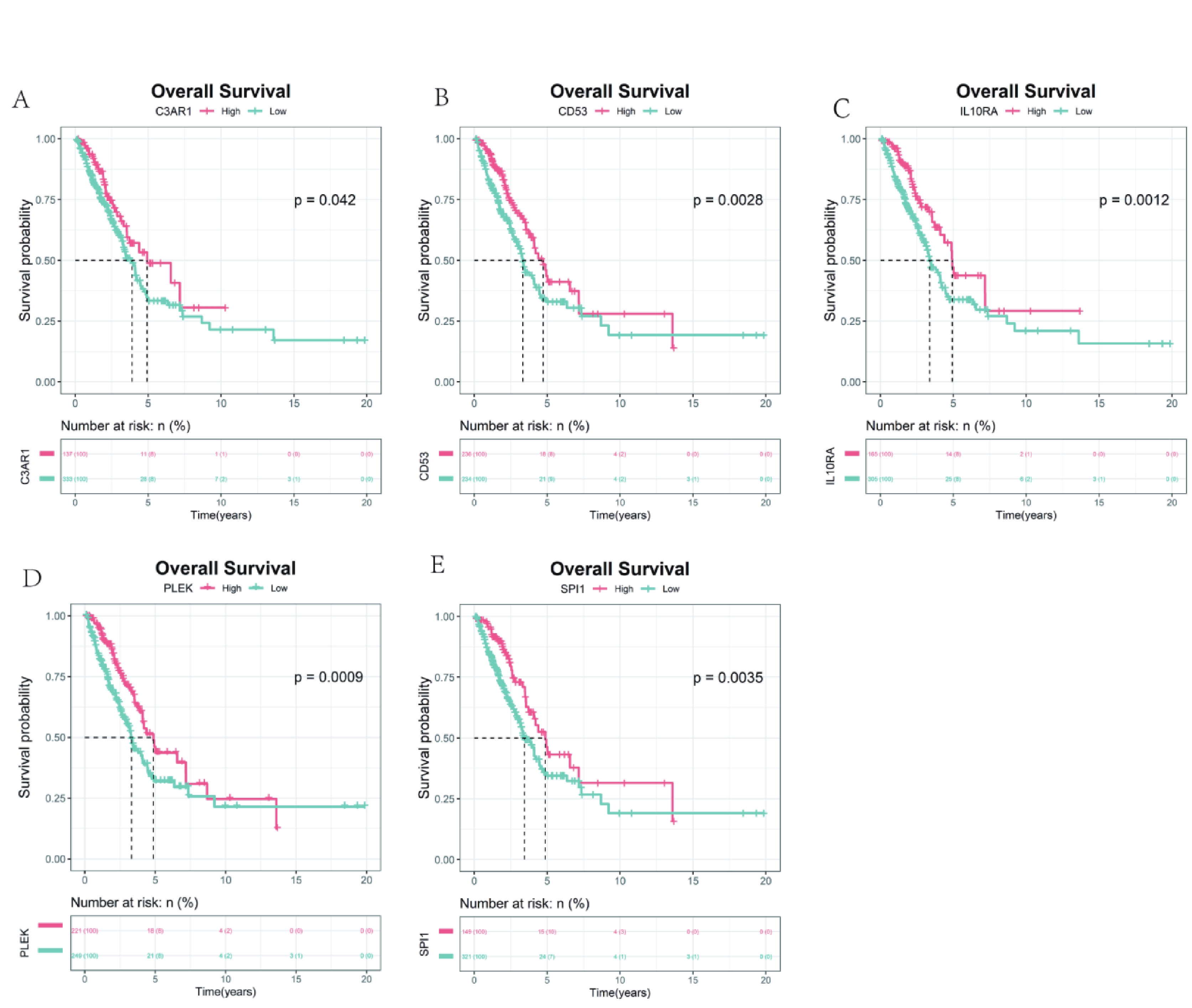 圖6
5個hub基因的Kaplan-Meier生存分析
圖6
5個hub基因的Kaplan-Meier生存分析
a~e:C3AR1,CD53,IL10RA,PLEK 以及 SPI1C3AR1的Kaplan-Meier生存分析
2.5 分析hub 基因與免疫細胞和免疫檢查點的相關性
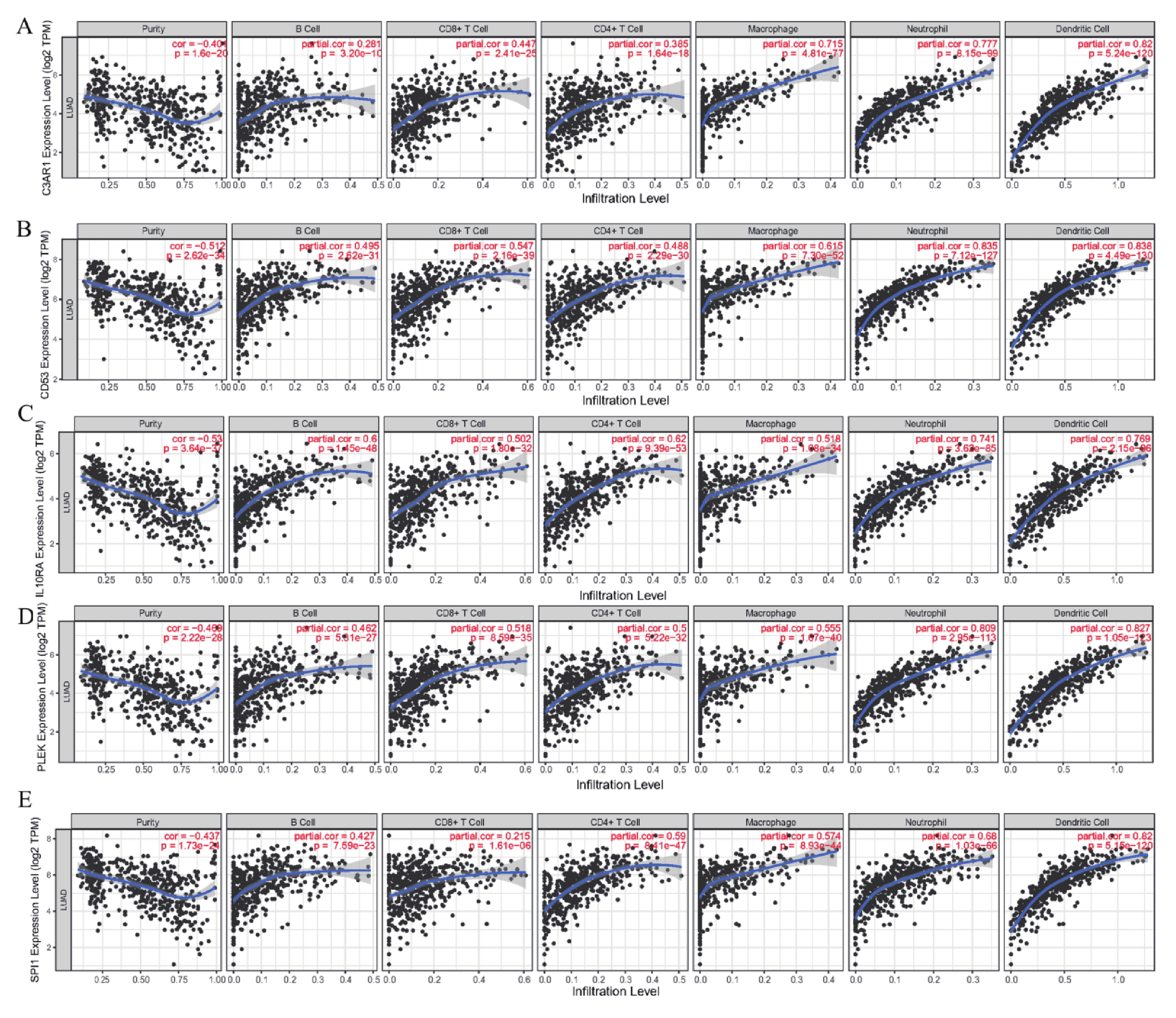
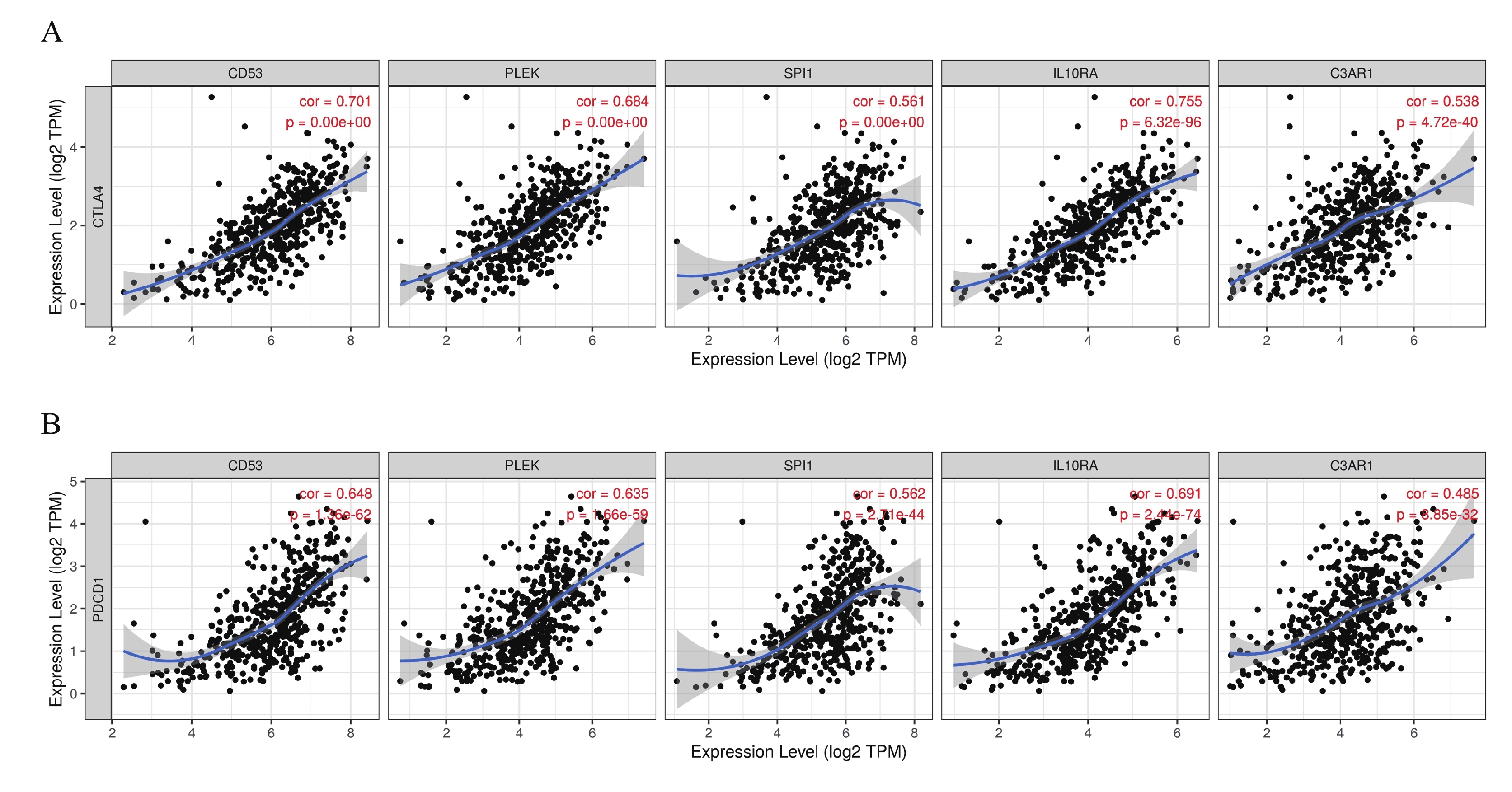
為了進一步分析5個hub基因與免疫細胞和免疫檢查點的相關性,通過TIMER網站對5個hub 基因進行評估。結果表明,這5個hub 基因的表達水平與6種免疫細胞的浸潤豐度均呈現顯著正相關,與腫瘤純度呈現顯著負相關(圖7)。同時,與免疫檢查點的相關性分析表明這5個hub 基因的表達水平與免疫檢查點CTLA4和PDCD1的表達量同樣呈現顯著正相關(圖8)。
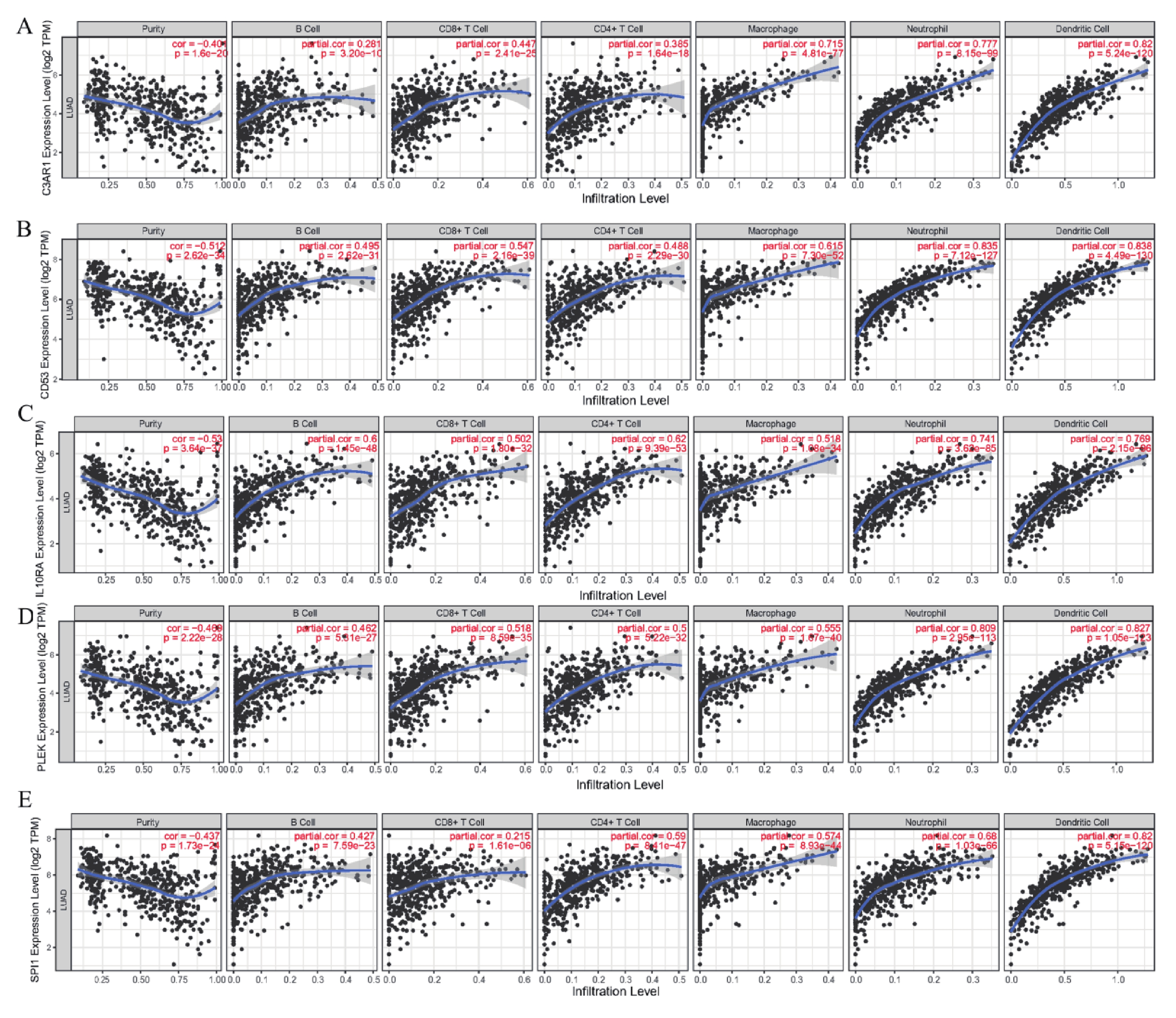 圖7
5個hub基因與免疫細胞及腫瘤純度的相關性分析
圖7
5個hub基因與免疫細胞及腫瘤純度的相關性分析
a~e:C3AR1,CD53,IL10RA,PLEK 以及 SPI1C3AR1與免疫細胞和腫瘤純度的相關性;X軸表示免疫細胞的浸潤水平和腫瘤純度,Y軸表示基因表達水平
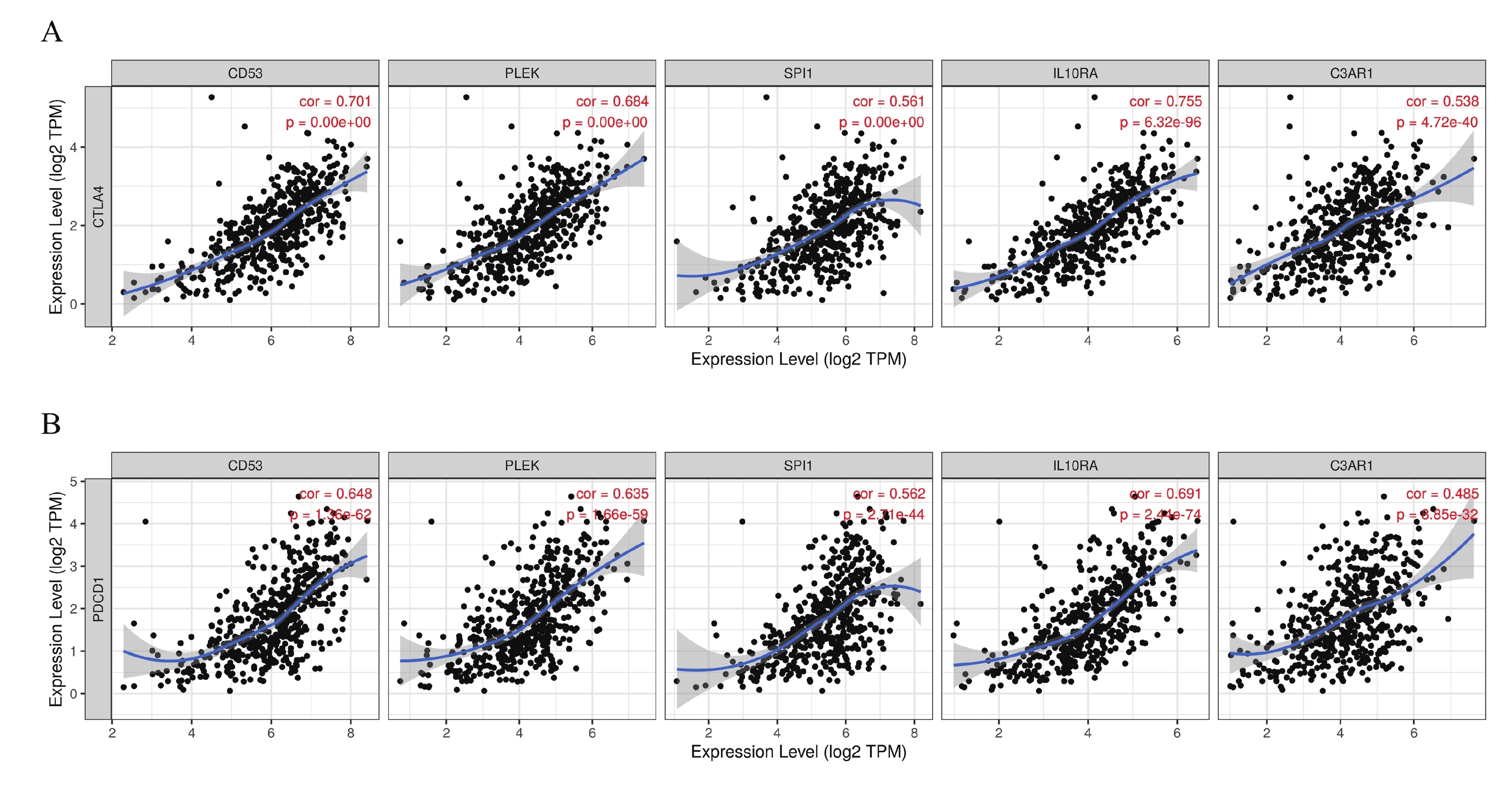 圖8
5個hub基因與免疫檢查點(CTLA4和PDCD1)的相關性分析
圖8
5個hub基因與免疫檢查點(CTLA4和PDCD1)的相關性分析
a:5個hub基因與CTLA4的相關性研究;b:5個hub基因與PDCD1的相關性研究
2.6 Hub 基因的GSEA富集分析
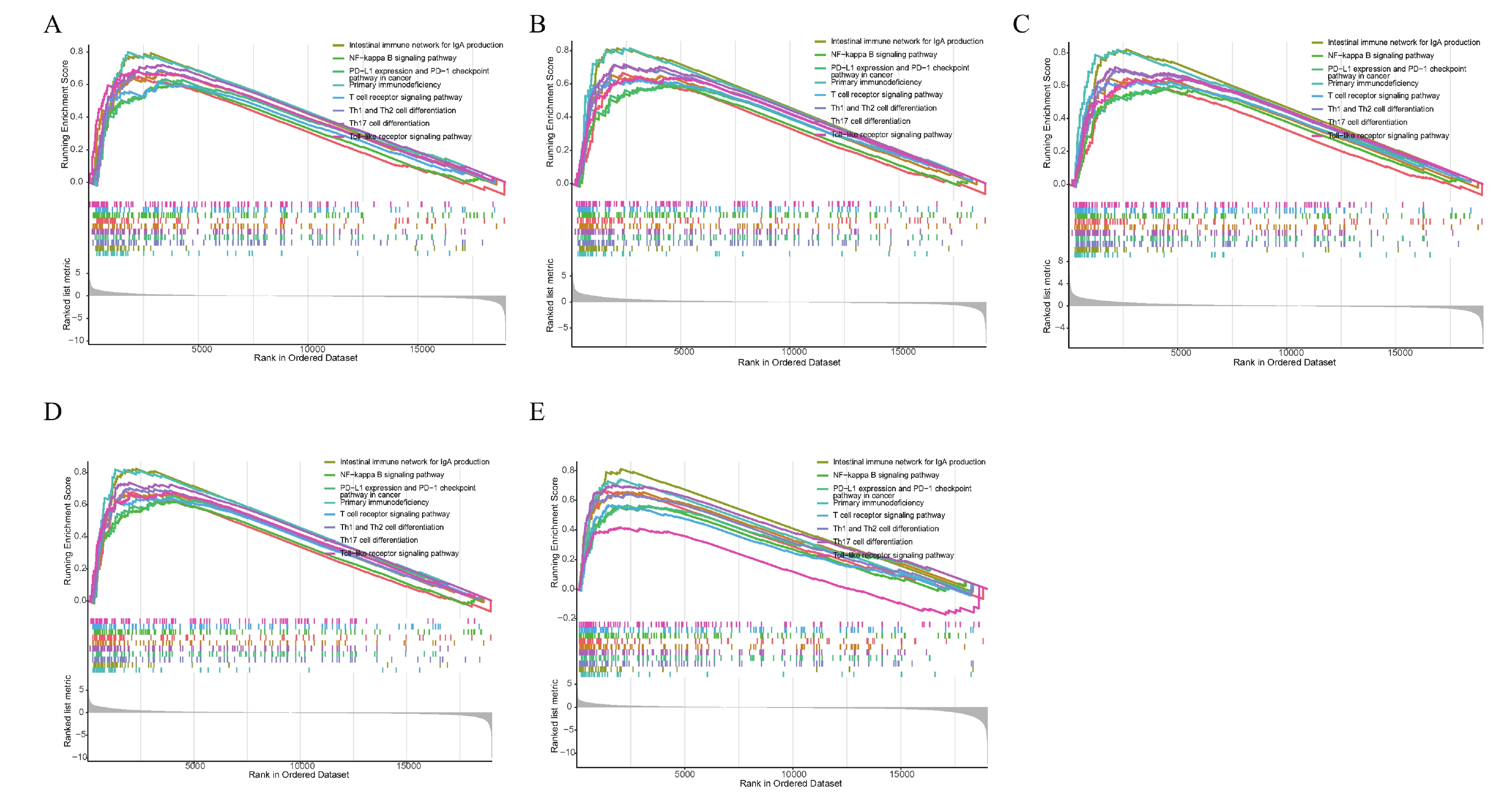
對5個hub 基因的高低表達組進行GSEA富集分析,結果表明,5個hub 基因均富集到Primary immunodeficiency、PD-L1 expression and PD-1 checkpoint pathway in cancer、NF-kappa B signaling pathway和Toll-like receptor signaling pathway等免疫相關的通路(圖9)。
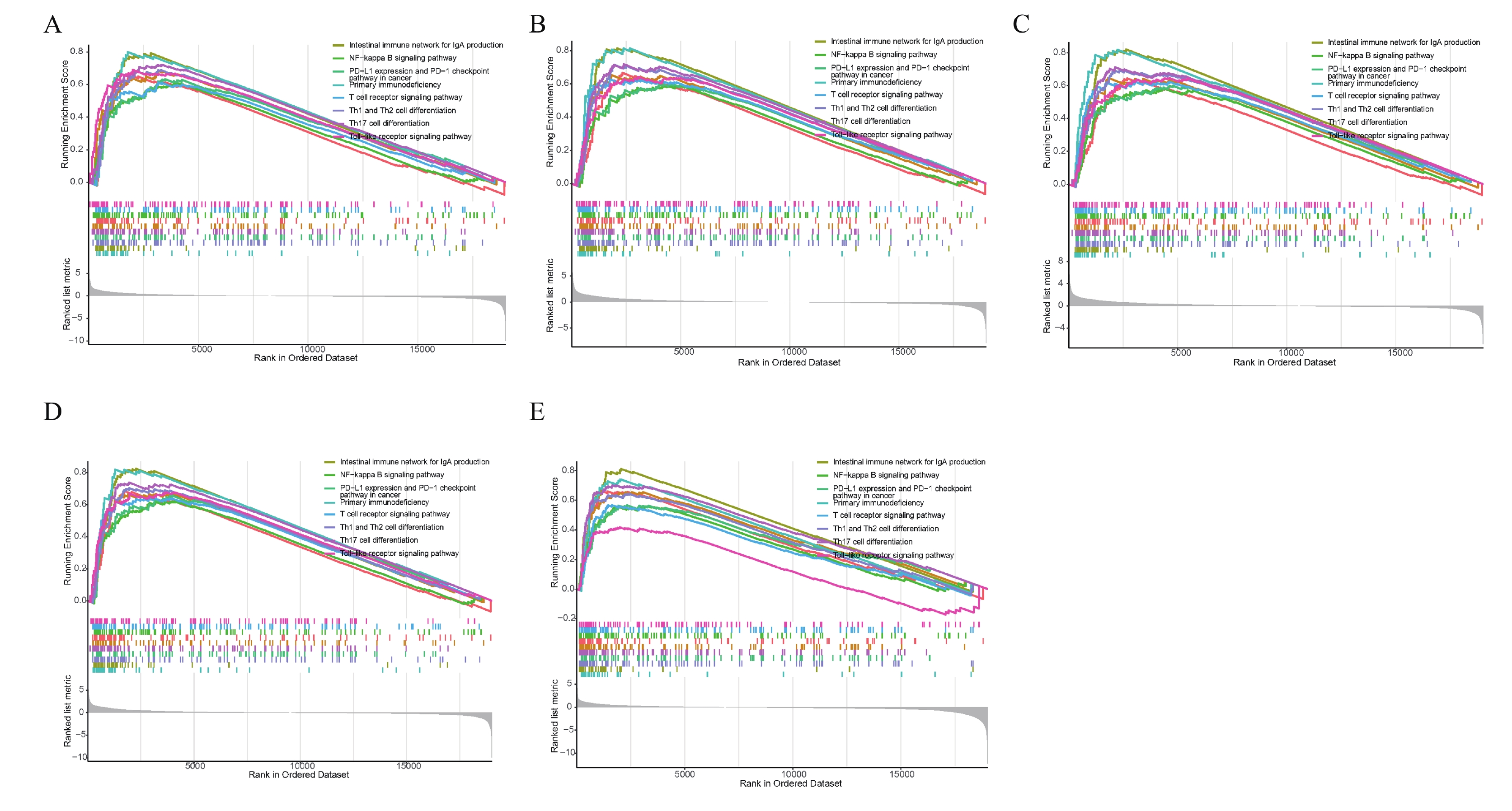 圖9
5個關鍵基因的GSEA富集分析
圖9
5個關鍵基因的GSEA富集分析
a~e:C3AR1,CD53,IL10RA,PLEK 以及 SPI1C3AR1的GSEA富集分析。上圖為通路的富集分數折線,下圖中的線對應各通路的基因
2.7 多因素cox回歸分析和生存分析
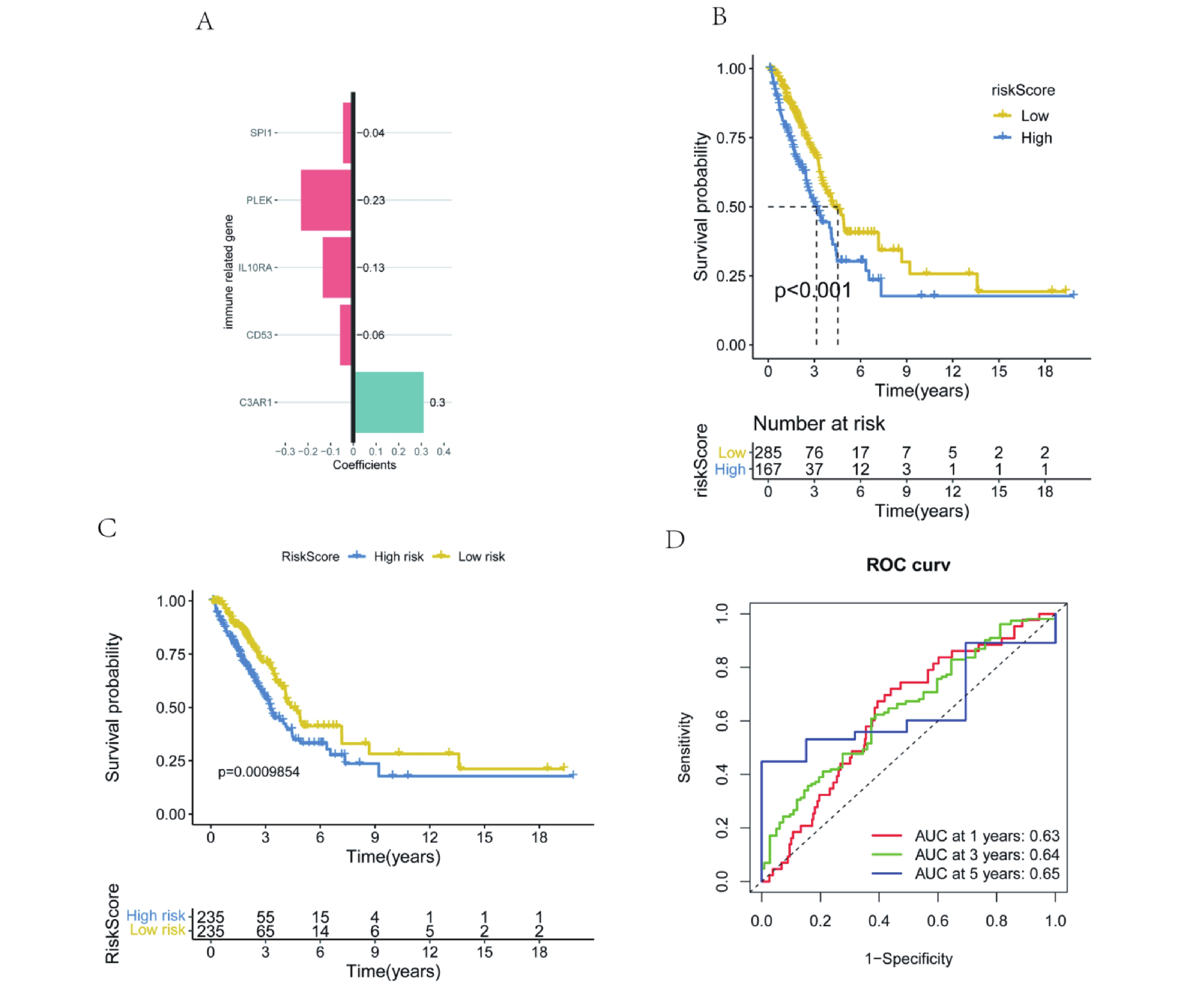
基于5個免疫相關的hub 基因進行多因素cox回歸分析并構建了預后風險模型,RiskScore=(?0.04×ExpressionSPI1)+(?0.23×ExpressionPLEK)+(?0.13×ExpressionIL10RA)+(?0.06×ExpressionCD53)+(0.3×ExpressionC3AR1)(圖10a)。同時,對患者的高低風險組進行了Kaplan-Meier生存分析。結果顯示,高風險組的患者預后較差(圖10b)。隨后,使用外部數據集GSE72094驗證模型,ROC曲線結果顯示,1年、3年和5年生存期的AUC值分別為0.63、0.64和0.65(圖10d)。生存曲線顯示,高風險組和低風險組的生存狀態存在顯著差異,高風險組的預后較差(圖10c)。以上結果證明,由5個特征基因構建的模型具有合理性。
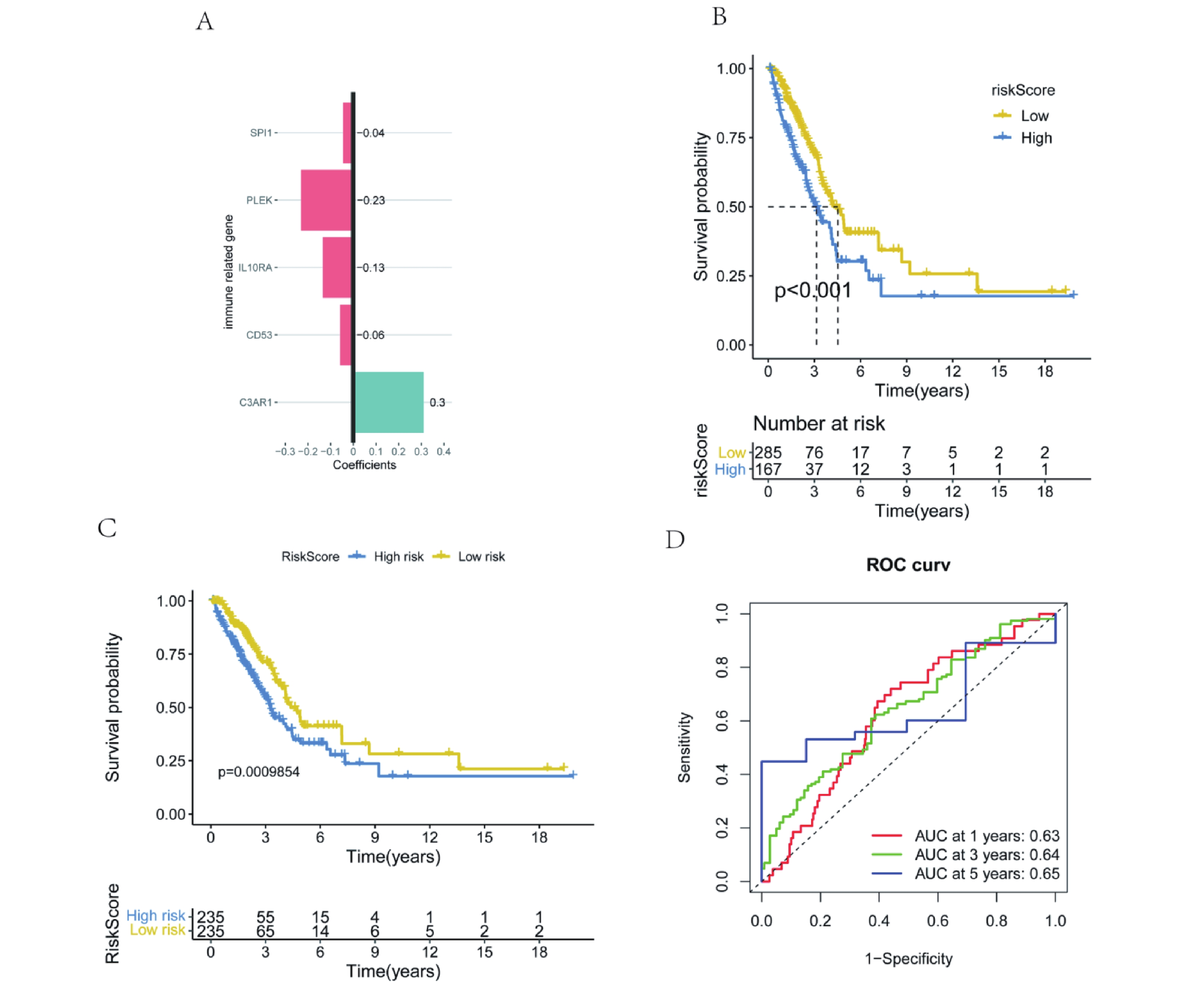 圖10
5個hub基因的多因素Cox回歸分析和 Kaplan-Meier生存分析
圖10
5個hub基因的多因素Cox回歸分析和 Kaplan-Meier生存分析
a:多因素Cox回歸分析森林圖;b:高風險和低風險組的Kaplan-Meier生存分析;c:外部驗證集中高風險組和低風險組的Kaplan-Meier生存分析;d: 外部驗證集的ROC曲線
2.8 評估RiskScore對免疫治療益處的預測能力
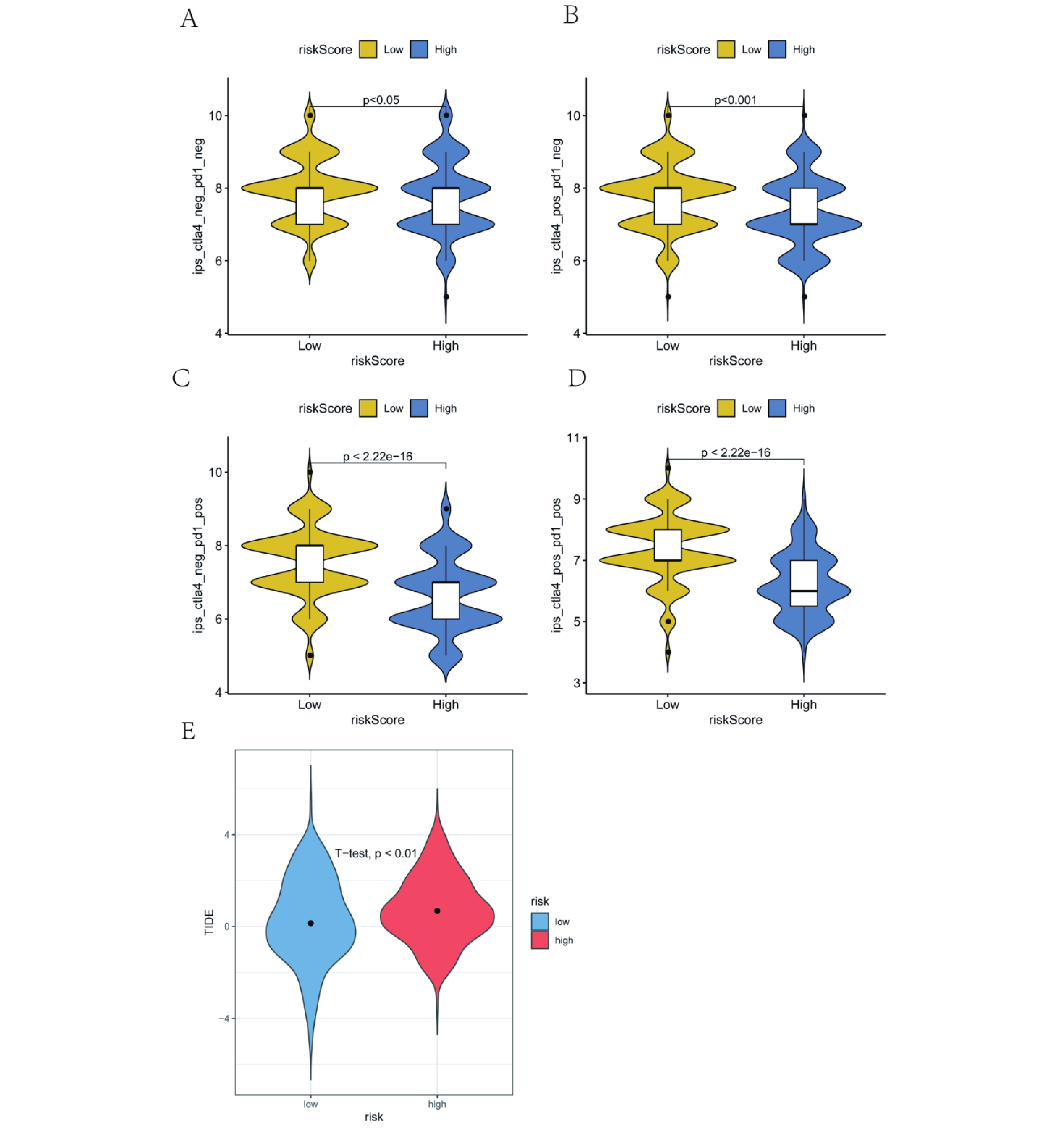
為了進一步分析RiskScore在預測治療益處中的作用,我們分析了高低風險組LUAD患者的IPS數據。IPS代表患者免疫原性,高IPS意味著患者有更高的免疫性能[19]。我們發現,IPS、IPS-CTLA4、IPS-PD1和IPS-CTLA4 and PD1在RiskScore組內差異具有統計學意義(P均<0.05)(圖11a~d)。先前的研究發現,TIDE可以準確預測患者的預后[20]。因此,我們計算了高低風險組患者的TIDE評分,結果顯示,低風險組的TIDE得分顯著低于高風險組(圖11e),這表明低風險組更能從免疫檢查點阻斷治療中受益。基于上述結果,我們認為我們建立的RiskScore在預測免疫治療方面有很大的潛力。
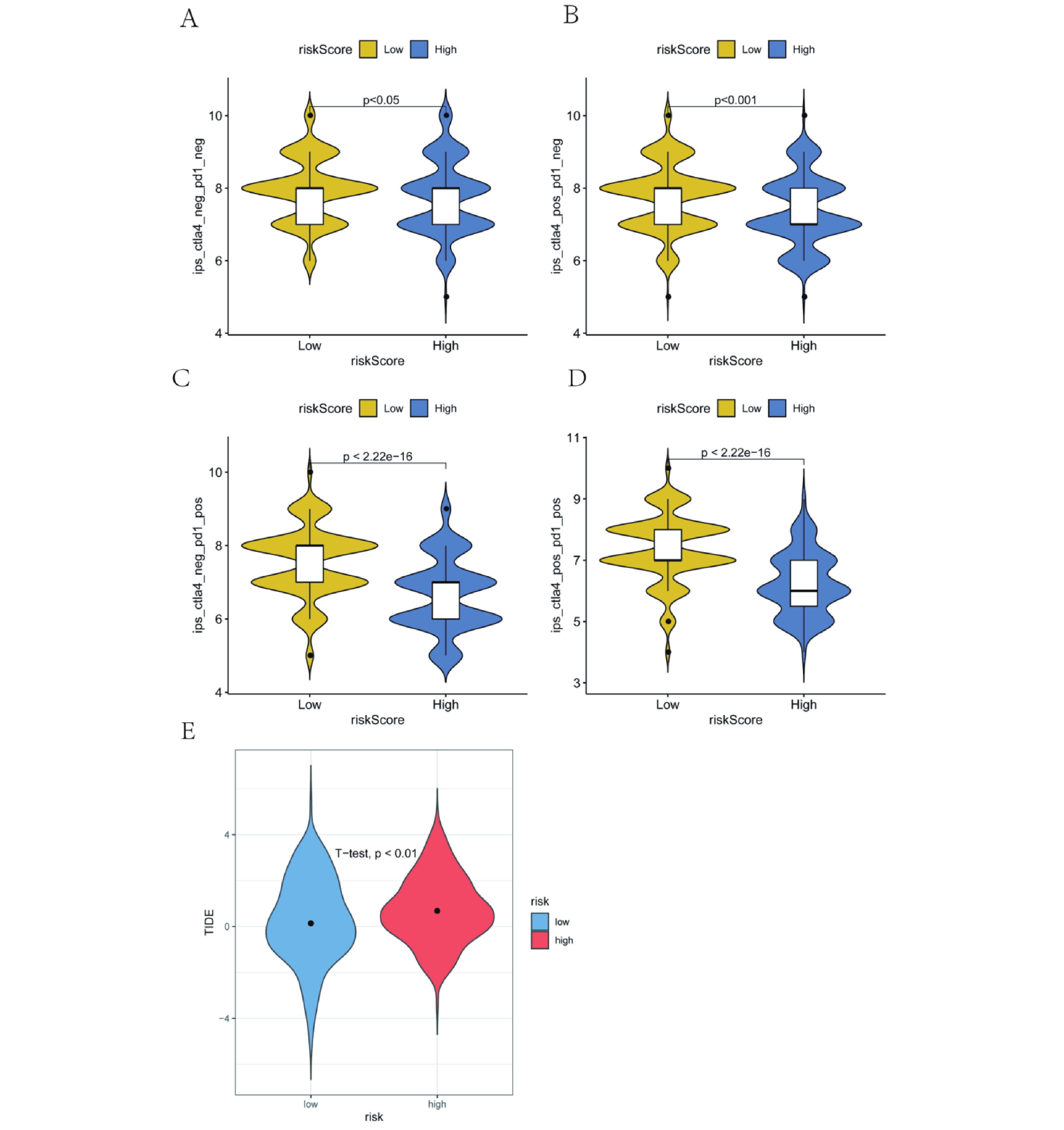 圖11
肺腺癌患者的IPS、TIDE和RiskScore之間的關系
圖11
肺腺癌患者的IPS、TIDE和RiskScore之間的關系
a~d:RiskScore組的IPS、IPS-CTLA4、IPS-PD1、IPS-CTL A4和PD1有顯著差異(均
3 討論
近年來,隨著診斷和治療方法的改進,LUAD的死亡率和發病率有了一定改善,但由于LUAD易轉移、易復發等原因導致該病患者的長期預后仍不令人滿意。目前,多項研究[21-22]發現腫瘤微環境的免疫細胞浸潤會影響腫瘤免疫治療的預后和臨床結果。因此,尋找免疫細胞浸潤相關關鍵基因為尋找有效診斷或治療LUAD的潛在靶點提供了一定的理論基礎。
本研究中,我們通過生存分析,發現5個hub基因的表達水平和LUAD患者的預后密切相關。同樣地,有研究[23]發現CD53的表達與三陰性乳腺癌患者的 無病生存期正相關,并且CD53在免疫細胞中表達,是淋巴細胞活化所必需的。相似地,有研究[24]發現CD53與骨肉瘤患者的生存時間顯著相關,并且在骨肉瘤轉移中發揮了重要作用,可以作為骨肉瘤轉移的候選生物標志物。Bai等[25]的研究發現,在LUAD中,PLEK是與腫瘤純度負相關的腫瘤純度共表達基因,并且可以作為預后風險因素預測LUAD患者的臨床結局。也有研究[26]表明SPI1誘導的lncRNA SNHG6上調可以通過miR-485-3p/VPS45軸促進前列腺癌進展。同時,Cheng等[27]的研究表明IL10RA的表達與黑色素瘤患者的預后顯著相關,并且體外實驗發現IL10RA能顯著抑制黑色素瘤細胞的增殖、遷移和侵襲,在黑色素瘤的進展中發揮了重要作用。Qu等[28]的研究發現,C3AR1與食管鱗狀細胞癌患者的生存時間相關,并且C3AR1可能通過影響巨噬細胞向M2表型的極化而導致免疫抑制微環境,從而進一步影響食管鱗狀細胞癌的進展,提示C3AR1可能是免疫治療的潛在靶點。綜上所述,這些研究表明我們獲得的hub 基因對腫瘤的發生發展和患者預后中都產生了重要影響。
多項研究[29-30]已證實,腫瘤的發生和進展會受到TME中免疫細胞和因子的調節,這些都可能會成為癌癥治療有希望的潛在靶點。在本研究中,我們發現5個hub 基因的表達水平與6種免疫細胞的浸潤豐度是顯著正相關的。已有研究[31-32]表明CD53是一種four-transmembrane protein,主要表達于骨髓淋巴系統,對B細胞功能是至關重要的,因為CD53促進BCR依賴性蛋白激酶C信號傳導,使其能夠磷酸化其底物,從而進一步促進B細胞活化。PLEK已被認為與免疫逃逸有關,并且是多種癌癥的潛在治療靶點[33]。SPI1原癌基因對于骨髓細胞、淋巴細胞和巨噬細胞的發育、成熟、分化和調控至關重要[34]。同時,研究[35]表明,SPI1的高表達與胃癌的免疫細胞浸潤相關,并參與細胞周期調控,導致預后不良。在膀胱癌中,SPI1可靶向調控CXCL12的表達,并募集腫瘤相關巨噬細胞[36]。IL10RA缺陷是原發性免疫缺陷病的病因[37]。IL10RA和IL10結合,出發JAK1-STAT3信號通路,抑制促炎細胞因子,還降低巨噬細胞活性,抑制單核細胞分化以及抗原呈遞細胞上共刺激分子的下調[38]。在癌癥中,IL10RA的抗炎和抗免疫反應也已經被報道多次[39-40]。C3AR1也在卵巢癌[41],腎透明細胞癌[42],骨肉瘤[43]等多種癌癥中發現與免疫細胞浸潤、介導腫瘤免疫微環境有關。由此可見,我們獲得的5個hub基因與免疫細胞的功能及激活密切相關,可以作為免疫細胞相關的潛在生物標志物進一步應用于LUAD的臨床免疫治療。
值得注意的是,我們對5個hub 基因進行了GSEA富集分析,結果發現這5個hub基因主要富集到Intestinal immune network for IgA production、Th1 and Th2 cell differentiation、PD-L1 expression and PD-1 checkpoint pathway in cancer、Th17 cell differentiation、NF-kappa B signaling pathway、T cell receptor signaling pathway和Toll-like receptor signaling pathway等免疫相關的通路。Yang等[44]的研究發現Bufalin可以通過APOBEC3F誘導的intestinal immune network for IgA production 信號通路抑制HCC細胞的增殖和遷移。Liu等[45]的研究發現,用從草珊瑚中純化的酸性多糖處理樹突狀細胞后,會使Th1 and Th2 cell differentiation通路中的DLL4基因顯著上調,從而進一步增強抗腫瘤免疫。Sun等[46]的研究表明,腫瘤外泌體傳遞的CRNDE-h通過抑制結直腸癌中Itch介導的泛素化和 RORγt降解來促進Th17 cell differentiation,從而進一步影響結直腸癌的發生和發展。Jin等[47]的研究則發現,TRIM7可以通過負調控NF-kappa B 信號通路顯著抑制肺癌細胞的增殖和遷移,為肺癌的治療提供了潛在靶點。同時,有研究[48]報道Toll-like receptor signaling pathway參與激活先天性和適應性免疫反應,在炎癥誘導的疾病中起著關鍵作用,該信號通路的失調可導致上皮層止血紊亂、慢性炎癥、過度修復反應和結直腸癌的發生。這些結果提示本研究所獲得的這5個hub基因可能會通過其相關的免疫信號通路在LUAD的發生和發展中發揮重要作用。
多項研究[49-50]證實,免疫治療為癌癥患者帶來了新的希望,尤其是免疫檢查點抑制劑的發現和應用在癌癥免疫治療中發揮了不可或缺的作用。如PD1 和 CTLA4等抑制劑有廣泛而多樣的機會通過調節免疫反應來增強抗腫瘤免疫[51]。在本研究中,我們發現基于5個hub 基因所構建的預后風險模型在預測免疫治療方面有很大的潛力,這為臨床實踐中治療不同風險患者的免疫治療策略的發展鋪墊了道路。在本研究中發現,低風險組的患者更可能從免疫檢查點治療當中獲益,這是基于理論層次上的研究。在實際情況當中,由于高風險患者的病情更為緊迫,病情更為復雜,醫生更傾向于盡快解決高風險患者的危急情況,穩定患者病情[52]。而對低風險的患者,盡管理論上他們更可能從治療當中獲益,在實際治療當中也應該考慮到藥物帶來的副作用和成本[53]。
綜上所述,我們的研究從理論上揭示了與免疫細胞密切相關的5個樞紐基因的巨大潛力,其不僅可以被選為LUAD免疫治療的預后生物標志物,而且可以被選作LUAD免疫療法的治療靶點。然而,本研究也存在一定的不足之處,本研究的結果均屬于理論層次上的研究,缺少臨床樣本和相關實驗的驗證。因此,對于使用外部驗證集驗證出hub基因的診斷效果不是十分理想,后期通過相關實驗對本研究的結果進行進一步驗證是必要的。
利益沖突:無。
作者貢獻:何東元負債設計并實施研究,分析數據,撰寫文章;陳波提供研究思路,分析研究方案可行性,并參與文章的修訂與審核;梁靖瑤參與提出文章的撰寫思路,收集并分析數據;葉海波:收集并分析數據,設計論文框架,并參與文章的修訂與終版文章的審核;易小杏、梁廣妮收集并分析數據,并參與文章的修訂;
肺癌是21世紀全球發生率和死亡率最高的惡性腫瘤,每年約有209萬新發病例和176萬死亡病例,嚴重威脅著人類生命健康[1-2]。其中,肺腺癌(lung adenocarcinoma,LUAD)是最普遍的組織病理學類型,發病率逐年上升[3]。目前,手術切除仍是治療LUAD最有效的方法。雖然隨著治療方法的多元化發展,晚期肺癌患者的生存狀況顯著改善,然而,其5年總生存率依然低于20%[4]。腫瘤微環境是由腫瘤、基質和內皮細胞組成的異質、復雜的組織,其特點是腫瘤與先天性和適應性免疫細胞之間的交互作用[5]。同時,研究[6]發現免疫微環境可通過促進腫瘤血管生成或調節腫瘤干細胞活性等方式直接或間接影響腫瘤的發生、發展。近年來,免疫治療作為一種新興治療模式,在LUAD治療中的潛力較大。目前針對免疫檢查點PD1/PD-L1、CTLA4的抗體已經廣泛應用于LUAD的治療[7]。同時,隨著免疫治療的進展,已有多項臨床試驗CheckMate-032 (NCT01928394)[8]、KEYNOTE-010(NCT01905657)[9]證實了Nivolumab、Pembrolizumab、Atezolizumab 較化療均顯著延長患者總生存期。然而,LUAD的免疫治療仍存在許多困境,如耐藥性增多、不良反應頻發、特異性低、療效出現瓶頸等問題,阻礙了其進一步發展。因此,挖掘安全有效的免疫相關的潛在作用靶點對于改善LUAD免疫治療困境來說具有重要意義。
近年來,利用生物信息學分析腫瘤免疫微環境,挖掘腫瘤生物標志物是一種流行而有效的方法。例如,利用WGCNA對乳腺癌進展和預后的研究[10]表明,有一個基因模塊與患者生存時間相關,并且該研究通過共表達網絡分析確定了8個候選生物標志物,為進一步了解乳腺癌的分子發病機制提供了依據。也有研究[11]通過ssGSEA將The Cancer Genome Atlas(TCGA)數據庫中舌鱗狀細胞癌患者分類為不同免疫細胞侵蝕的集群,并利用ESTIMATE和CIBERSORT分析對集群之間的免疫狀況進行評估,最終確定了15個免疫相關基因可以作為舌鱗狀細胞癌患者預后的潛在生物標志物。由此可見,通過生物信息學的分析方法挖掘癌癥治療的潛在生物標志物已受到眾多研究者的關注。
本研究通過WGCNA和ESTIMATE等生物信息學分析方法對TCGA數據庫來源的LUAD表達數據進行分析,最終獲得與免疫細胞相關的hub基因。同時,基于hub基因構建了預后風險模型,并評估了RiskScore對免疫治療益處的預測能力。總的來說,本研究的結果有利于尋找LUAD潛在的診斷或治療生物標志物,這對于臨床為患者提供個性化的治療策略具有重要意義。
1 資料與方法
1.1 數據來源
LUAD mRNA表達數據集(FPKM)TCGA-LUAD和對應的臨床信息從TCGA數據庫(
1.2 肺腺癌樣本免疫微環境分析
利用R包“estimate”[12]對TCGA-LUAD數據集中LUAD腫瘤樣本腫瘤微環境進行評估。
1.3 加權基因共表達網絡分析
首先通過R包“WGCNA”來建立基因共表達網絡[13]。然后通過使用 Pearson 相關函數測量 TCGA-LUAD 數據中基因之間的共表達。通過將共表達相似度提高到β=5來構建鄰接矩陣。從鄰接矩陣出發,構建拓撲重疊矩陣(TOM)來考慮拓撲相似性,并建立相應的不相似矩陣(1-TOM)來形成聚類。使用hclust函數利用不同矩陣進行分層聚類。最后基于拓撲重疊矩陣進行層次聚類,按照動態混合切割方法,每個模塊最少基因數目設置為50,相異性閾值設置為0.25。計算由第一主成分(PC1)表示的模塊特征基因表達式(ME),確定每個模塊的表達式。臨床資料與模塊特征基因表達相關,并確定基因顯著性(gene significance,GS)。通過將模塊的ME與模塊的基因表達相關聯,確定模塊成員的定量方法,顏色被隨機分配給模塊,最終選擇與免疫評分、基質評分和ESTIMATE評分顯著相關的模塊作為后續的研究對象。為了進一步探究特征模塊內基因的生物學功能,利用R包“clusterProfiler”[14]對特征模塊內基因進行富集分析,特征模塊基因最顯著富集terms的篩選標準為P<0.05和Q<0.05。
1.4 獲取hub基因
本研究以cor.geneModuleMembership>0.8 和cor.geneTraitSignificance>0.6 為閾值篩選模塊內候選關鍵基因。同時,利用特征模塊內的所有基因在STRING (http://string-db.org/cgi/input.pl)網站構建 蛋白質-蛋白質相互作用(PPI)網絡,以相互作用評分>0.4 作為閾值,將獲得的候選hub基因與PPI 網絡中degree前30的基因取交集,最終獲得hub 基因。
1.5 hub基因的進一步驗證
首先比較hub基因在正常組中的表達差異。然后通過Pearson相關性分析hub 基因與各Stage分期之間的關系。Cancer Cell Line Encyclopedia(CCLE)數據庫(
1.6 評估RiskScore對免疫治療益處的預測能力
為了進一步分析RiskScore在預測治療益處中的作用。我們在獲得LUAD患者IPS數據的基礎上,將患者按風險評分的中位值分為高風險組和低風險組,利用t檢驗比較anti-PD1和anti-CTLA4 的治療效果在高低風險組中的差異(P<0.05),并利用R包“ggpubr”(
2 結果
2.1 WGCNA和特征模塊的識別
利用R包“WGCNA”對預處理后的511例腫瘤樣本MAD值最大的前5 000個基因構建加權基因共表達網絡。本研究中是以軟閾值為β=5來建立無尺度化網絡(圖1a~d)。接下來,基于平均層次聚類和動態樹切割,最終共獲得13個基因模塊(圖1e,表1)。同時,13個基因模塊和臨床性狀的相關性分析結果表明,brown模塊與StromalScore(r=0.67,P=6e-68)、ImmuneScore(r=0.91,P=1e-198)、ESTIMATEScore(r=0.86,P =3e-153)呈顯著正相關,與TumorPurity呈負相關(r=–0.88,P=3e-169)(圖1f),因此我們假設用于構建brown模塊的基因可能與免疫力有關。我們選擇brown模塊作為后續分析的研究對象。
圖1
肺腺癌的加權基因共表達網絡
a:不同軟閾值功率下的無標度擬合指數分析(β);X軸表示軟閾值,Y軸表示不同軟閾值下的無標度擬合指數;紅線是對應于WGCNA分析的無標度擬合指數,最優軟閾值為5;b:不同軟閾值功率下的平均連通性;X軸表示軟閾值功率,Y軸反映平均連通性,表示不同軟閾值下的網絡連接性;c:β=5時連通性分布直方圖;d:檢查β=5時的無標度拓撲;e:基于不同度量(1-TOM)聚類的所有差異表達基因的樹狀圖,共13個不同的模塊,不同的顏色作為標識符標識不同的模塊;f:熱圖表示模塊hub基因與臨床性狀之間的相關性。ME表示模塊本征基因,它是在對模塊中的所有基因進行PCA分析后獲得的主成分1(PC1)的值;紅色表示與表型呈正相關,藍色表示與表型負相關
2.2 特征模塊內基因的生物功能和通路富集分析
對brown模塊內的521個基因進行富集分析,結果表明該模塊內基因大多與中性粒細胞活化參與免疫反應、先天免疫反應的調節、細胞激活的正向調節、中性粒細胞活化和白細胞增殖等免疫功能相關;與lumenal side of endoplasmic reticulum membrane、integral component of lumenal side of endoplasmic reticulum membrane等內質網細胞組分相關;與MHC class Ⅱ protein complex binding和MHC class Ⅱ receptor activity等MHC受體活性有關(圖2a)。KEGG富集分析結果顯示該模塊內基因主要富集到NF-kappa B signaling pathway、NOD-like receptor signaling pathway、primary immunodeficiency、complement and coagulation cascade和human T-cell leukemia virus 1 infection等免疫和癌癥相關通路中(圖2b)。上述結果表明brown模塊中的基因主要與免疫功能相關,并在免疫相關途徑中富集。
圖2
Brown模塊基因的富集分析
a:brown模塊中hub基因的GO功能富集分析,縱坐標表示不同的GO項,橫坐標表示基因富集數;b:brown模塊特征基因KEGG通路富集分析,縱坐標為KEGG通路注釋,橫坐標為基因富集數。 圖中的點的大小表示富集基因的數量;
2.3 與免疫相關的hub基因的鑒定
在brown基因模塊中篩選出與ImmuneScore具有較高相關性的27個基因作為候選hub 基因(圖3a)。同時,將brown模塊中的所有基因輸入STRING網站并構建PPI網絡,根據degree值篩選出前30基因(圖3b)。以相互作用評分>0.4作為閾值,將27個候選基因與前30基因取交集,最終獲得5個hub 基因,分別為CD53、PLEK、SPI1、IL10RA和C3AR1(圖3c)。
圖3
與免疫相關的hub基因的鑒定
a:散點圖表示brown模塊中與組織學免疫評分相關的hub基因,X軸值表示基因和模塊之間的相關性;如果絕對值接近0,則該基因不是該模塊的一部分;如果這個值的絕對值接近1,那么該基因與該模塊高度相關。Y軸值表示基因和免疫評分之間的相關性;0表示該基因與免疫力不相關,1表示該基因與免疫力高度相關,圖中每個點代表一個基因;b:brown模塊中PPI網絡節點的前30個基因;c:韋恩圖篩選最終與免疫相關的關鍵基因;27個候選hub基因與PPI中degree前30個基因取交集
2.4 正常組與腫瘤組hub基因的表達差異及其與腫瘤分期的相關性及hub基因的Kaplan-Meier生存分析
首先利用t檢驗分析了5個hub基因在正常組和腫瘤組中的表達差異,結果顯示,與正常組相比,5個hub 基因在腫瘤組中顯著低表達(圖4a~e)。同時,分析了5個hub基因的表達與Stage分期之間的相關性,結果顯示, CD53和IL10RA的表達水平與Stage分期有關聯(圖4f)。CCLE數據庫有大量的人類癌癥細胞系和獨特的數據集,能夠對大量人類癌癥細胞系進行詳細的遺傳表征[18]。因此,為了探索關鍵基因在大量LUAD細胞系中的表達差異,我們利用CCLE數據庫發現SPI1、PLEK、IL10RA、CD53和C3AR1在LUAD細胞株中的表達存在差異(圖5)。同時對患者的高低表達組進行生存分析,結果發現5個hub 基因的高表達預示著患者較好的預后(圖6)。
圖4
分析5個關鍵基因在正常組和腫瘤組的表達差異及在不同腫瘤分期中的mRNA表達變化
a~e:正常組和腫瘤組中C3AR1、CD53、IL10RA、PLEK和SPI1表達的差異;f:箱線圖表示5個hub基因在不同腫瘤分期中的mRNA表達情況。
圖5
5個hub基因在大量肺腺癌細胞系中的表達
通過CCLE分析肺腺癌細胞系中SPI1(a)、PLEK(b)、IL10RA(c)、CD53(d)和C3AR1(e)的表達水平;紅色表示高表達,藍色表示低表達
圖6
5個hub基因的Kaplan-Meier生存分析
a~e:C3AR1,CD53,IL10RA,PLEK 以及 SPI1C3AR1的Kaplan-Meier生存分析
2.5 分析hub 基因與免疫細胞和免疫檢查點的相關性
為了進一步分析5個hub基因與免疫細胞和免疫檢查點的相關性,通過TIMER網站對5個hub 基因進行評估。結果表明,這5個hub 基因的表達水平與6種免疫細胞的浸潤豐度均呈現顯著正相關,與腫瘤純度呈現顯著負相關(圖7)。同時,與免疫檢查點的相關性分析表明這5個hub 基因的表達水平與免疫檢查點CTLA4和PDCD1的表達量同樣呈現顯著正相關(圖8)。
圖7
5個hub基因與免疫細胞及腫瘤純度的相關性分析
a~e:C3AR1,CD53,IL10RA,PLEK 以及 SPI1C3AR1與免疫細胞和腫瘤純度的相關性;X軸表示免疫細胞的浸潤水平和腫瘤純度,Y軸表示基因表達水平
圖8
5個hub基因與免疫檢查點(CTLA4和PDCD1)的相關性分析
a:5個hub基因與CTLA4的相關性研究;b:5個hub基因與PDCD1的相關性研究
2.6 Hub 基因的GSEA富集分析
對5個hub 基因的高低表達組進行GSEA富集分析,結果表明,5個hub 基因均富集到Primary immunodeficiency、PD-L1 expression and PD-1 checkpoint pathway in cancer、NF-kappa B signaling pathway和Toll-like receptor signaling pathway等免疫相關的通路(圖9)。
圖9
5個關鍵基因的GSEA富集分析
a~e:C3AR1,CD53,IL10RA,PLEK 以及 SPI1C3AR1的GSEA富集分析。上圖為通路的富集分數折線,下圖中的線對應各通路的基因
2.7 多因素cox回歸分析和生存分析
基于5個免疫相關的hub 基因進行多因素cox回歸分析并構建了預后風險模型,RiskScore=(?0.04×ExpressionSPI1)+(?0.23×ExpressionPLEK)+(?0.13×ExpressionIL10RA)+(?0.06×ExpressionCD53)+(0.3×ExpressionC3AR1)(圖10a)。同時,對患者的高低風險組進行了Kaplan-Meier生存分析。結果顯示,高風險組的患者預后較差(圖10b)。隨后,使用外部數據集GSE72094驗證模型,ROC曲線結果顯示,1年、3年和5年生存期的AUC值分別為0.63、0.64和0.65(圖10d)。生存曲線顯示,高風險組和低風險組的生存狀態存在顯著差異,高風險組的預后較差(圖10c)。以上結果證明,由5個特征基因構建的模型具有合理性。
圖10
5個hub基因的多因素Cox回歸分析和 Kaplan-Meier生存分析
a:多因素Cox回歸分析森林圖;b:高風險和低風險組的Kaplan-Meier生存分析;c:外部驗證集中高風險組和低風險組的Kaplan-Meier生存分析;d: 外部驗證集的ROC曲線
2.8 評估RiskScore對免疫治療益處的預測能力
為了進一步分析RiskScore在預測治療益處中的作用,我們分析了高低風險組LUAD患者的IPS數據。IPS代表患者免疫原性,高IPS意味著患者有更高的免疫性能[19]。我們發現,IPS、IPS-CTLA4、IPS-PD1和IPS-CTLA4 and PD1在RiskScore組內差異具有統計學意義(P均<0.05)(圖11a~d)。先前的研究發現,TIDE可以準確預測患者的預后[20]。因此,我們計算了高低風險組患者的TIDE評分,結果顯示,低風險組的TIDE得分顯著低于高風險組(圖11e),這表明低風險組更能從免疫檢查點阻斷治療中受益。基于上述結果,我們認為我們建立的RiskScore在預測免疫治療方面有很大的潛力。
圖11
肺腺癌患者的IPS、TIDE和RiskScore之間的關系
a~d:RiskScore組的IPS、IPS-CTLA4、IPS-PD1、IPS-CTL A4和PD1有顯著差異(均
3 討論
近年來,隨著診斷和治療方法的改進,LUAD的死亡率和發病率有了一定改善,但由于LUAD易轉移、易復發等原因導致該病患者的長期預后仍不令人滿意。目前,多項研究[21-22]發現腫瘤微環境的免疫細胞浸潤會影響腫瘤免疫治療的預后和臨床結果。因此,尋找免疫細胞浸潤相關關鍵基因為尋找有效診斷或治療LUAD的潛在靶點提供了一定的理論基礎。
本研究中,我們通過生存分析,發現5個hub基因的表達水平和LUAD患者的預后密切相關。同樣地,有研究[23]發現CD53的表達與三陰性乳腺癌患者的 無病生存期正相關,并且CD53在免疫細胞中表達,是淋巴細胞活化所必需的。相似地,有研究[24]發現CD53與骨肉瘤患者的生存時間顯著相關,并且在骨肉瘤轉移中發揮了重要作用,可以作為骨肉瘤轉移的候選生物標志物。Bai等[25]的研究發現,在LUAD中,PLEK是與腫瘤純度負相關的腫瘤純度共表達基因,并且可以作為預后風險因素預測LUAD患者的臨床結局。也有研究[26]表明SPI1誘導的lncRNA SNHG6上調可以通過miR-485-3p/VPS45軸促進前列腺癌進展。同時,Cheng等[27]的研究表明IL10RA的表達與黑色素瘤患者的預后顯著相關,并且體外實驗發現IL10RA能顯著抑制黑色素瘤細胞的增殖、遷移和侵襲,在黑色素瘤的進展中發揮了重要作用。Qu等[28]的研究發現,C3AR1與食管鱗狀細胞癌患者的生存時間相關,并且C3AR1可能通過影響巨噬細胞向M2表型的極化而導致免疫抑制微環境,從而進一步影響食管鱗狀細胞癌的進展,提示C3AR1可能是免疫治療的潛在靶點。綜上所述,這些研究表明我們獲得的hub 基因對腫瘤的發生發展和患者預后中都產生了重要影響。
多項研究[29-30]已證實,腫瘤的發生和進展會受到TME中免疫細胞和因子的調節,這些都可能會成為癌癥治療有希望的潛在靶點。在本研究中,我們發現5個hub 基因的表達水平與6種免疫細胞的浸潤豐度是顯著正相關的。已有研究[31-32]表明CD53是一種four-transmembrane protein,主要表達于骨髓淋巴系統,對B細胞功能是至關重要的,因為CD53促進BCR依賴性蛋白激酶C信號傳導,使其能夠磷酸化其底物,從而進一步促進B細胞活化。PLEK已被認為與免疫逃逸有關,并且是多種癌癥的潛在治療靶點[33]。SPI1原癌基因對于骨髓細胞、淋巴細胞和巨噬細胞的發育、成熟、分化和調控至關重要[34]。同時,研究[35]表明,SPI1的高表達與胃癌的免疫細胞浸潤相關,并參與細胞周期調控,導致預后不良。在膀胱癌中,SPI1可靶向調控CXCL12的表達,并募集腫瘤相關巨噬細胞[36]。IL10RA缺陷是原發性免疫缺陷病的病因[37]。IL10RA和IL10結合,出發JAK1-STAT3信號通路,抑制促炎細胞因子,還降低巨噬細胞活性,抑制單核細胞分化以及抗原呈遞細胞上共刺激分子的下調[38]。在癌癥中,IL10RA的抗炎和抗免疫反應也已經被報道多次[39-40]。C3AR1也在卵巢癌[41],腎透明細胞癌[42],骨肉瘤[43]等多種癌癥中發現與免疫細胞浸潤、介導腫瘤免疫微環境有關。由此可見,我們獲得的5個hub基因與免疫細胞的功能及激活密切相關,可以作為免疫細胞相關的潛在生物標志物進一步應用于LUAD的臨床免疫治療。
值得注意的是,我們對5個hub 基因進行了GSEA富集分析,結果發現這5個hub基因主要富集到Intestinal immune network for IgA production、Th1 and Th2 cell differentiation、PD-L1 expression and PD-1 checkpoint pathway in cancer、Th17 cell differentiation、NF-kappa B signaling pathway、T cell receptor signaling pathway和Toll-like receptor signaling pathway等免疫相關的通路。Yang等[44]的研究發現Bufalin可以通過APOBEC3F誘導的intestinal immune network for IgA production 信號通路抑制HCC細胞的增殖和遷移。Liu等[45]的研究發現,用從草珊瑚中純化的酸性多糖處理樹突狀細胞后,會使Th1 and Th2 cell differentiation通路中的DLL4基因顯著上調,從而進一步增強抗腫瘤免疫。Sun等[46]的研究表明,腫瘤外泌體傳遞的CRNDE-h通過抑制結直腸癌中Itch介導的泛素化和 RORγt降解來促進Th17 cell differentiation,從而進一步影響結直腸癌的發生和發展。Jin等[47]的研究則發現,TRIM7可以通過負調控NF-kappa B 信號通路顯著抑制肺癌細胞的增殖和遷移,為肺癌的治療提供了潛在靶點。同時,有研究[48]報道Toll-like receptor signaling pathway參與激活先天性和適應性免疫反應,在炎癥誘導的疾病中起著關鍵作用,該信號通路的失調可導致上皮層止血紊亂、慢性炎癥、過度修復反應和結直腸癌的發生。這些結果提示本研究所獲得的這5個hub基因可能會通過其相關的免疫信號通路在LUAD的發生和發展中發揮重要作用。
多項研究[49-50]證實,免疫治療為癌癥患者帶來了新的希望,尤其是免疫檢查點抑制劑的發現和應用在癌癥免疫治療中發揮了不可或缺的作用。如PD1 和 CTLA4等抑制劑有廣泛而多樣的機會通過調節免疫反應來增強抗腫瘤免疫[51]。在本研究中,我們發現基于5個hub 基因所構建的預后風險模型在預測免疫治療方面有很大的潛力,這為臨床實踐中治療不同風險患者的免疫治療策略的發展鋪墊了道路。在本研究中發現,低風險組的患者更可能從免疫檢查點治療當中獲益,這是基于理論層次上的研究。在實際情況當中,由于高風險患者的病情更為緊迫,病情更為復雜,醫生更傾向于盡快解決高風險患者的危急情況,穩定患者病情[52]。而對低風險的患者,盡管理論上他們更可能從治療當中獲益,在實際治療當中也應該考慮到藥物帶來的副作用和成本[53]。
綜上所述,我們的研究從理論上揭示了與免疫細胞密切相關的5個樞紐基因的巨大潛力,其不僅可以被選為LUAD免疫治療的預后生物標志物,而且可以被選作LUAD免疫療法的治療靶點。然而,本研究也存在一定的不足之處,本研究的結果均屬于理論層次上的研究,缺少臨床樣本和相關實驗的驗證。因此,對于使用外部驗證集驗證出hub基因的診斷效果不是十分理想,后期通過相關實驗對本研究的結果進行進一步驗證是必要的。
利益沖突:無。
作者貢獻:何東元負債設計并實施研究,分析數據,撰寫文章;陳波提供研究思路,分析研究方案可行性,并參與文章的修訂與審核;梁靖瑤參與提出文章的撰寫思路,收集并分析數據;葉海波:收集并分析數據,設計論文框架,并參與文章的修訂與終版文章的審核;易小杏、梁廣妮收集并分析數據,并參與文章的修訂;