

胃癌是高度惡性腫瘤,其死亡率居全球惡性腫瘤第5位[1]。我國胃癌發病率較高,發病率及死亡率均居惡性腫瘤的第3位[2]。近年來,在低風險和高風險國家,年輕人的胃癌發病率在逐步上升[3]。化療是轉移性胃癌(metastatic gastric cancer,mGC)的主要治療方法,對于接受常規化療的患者,中位總生存期(overall survival,OS)約為12個月[4]。免疫療法在胃癌治療方面的效果顯著,已成為晚期胃癌的一線治療方案[5]。免疫檢查點抑制劑(immune checkpoint inhibitors,ICIs)等新型免疫治療的出現在一定程度上改變了胃癌患者的治療。然而,只有少數晚期胃癌患者對ICIs治療有持久反應,且ICIs的療效非常有限[6]。腫瘤相關巨噬細胞(tumor-associated macrophage,TAM)在胃癌的免疫治療中發揮重要作用的同時,可能會導致免疫治療的耐藥性。這種耐藥性可能是由于TAM的免疫抑制作用,或者是由于它們在腫瘤微環境(tumor microenvironment,TME)中的功能受到干擾。因此,了解TAM在免疫治療中的作用和調控機制對于克服免疫治療耐藥性具有重要意義。筆者現就TAM的發生發展及其與癌癥的關系予以綜述,重點闡述TAM在胃癌免疫治療及耐藥方面可能存在的機制,以及針對耐藥可能的治療手段。
1 TAM的起源和分類
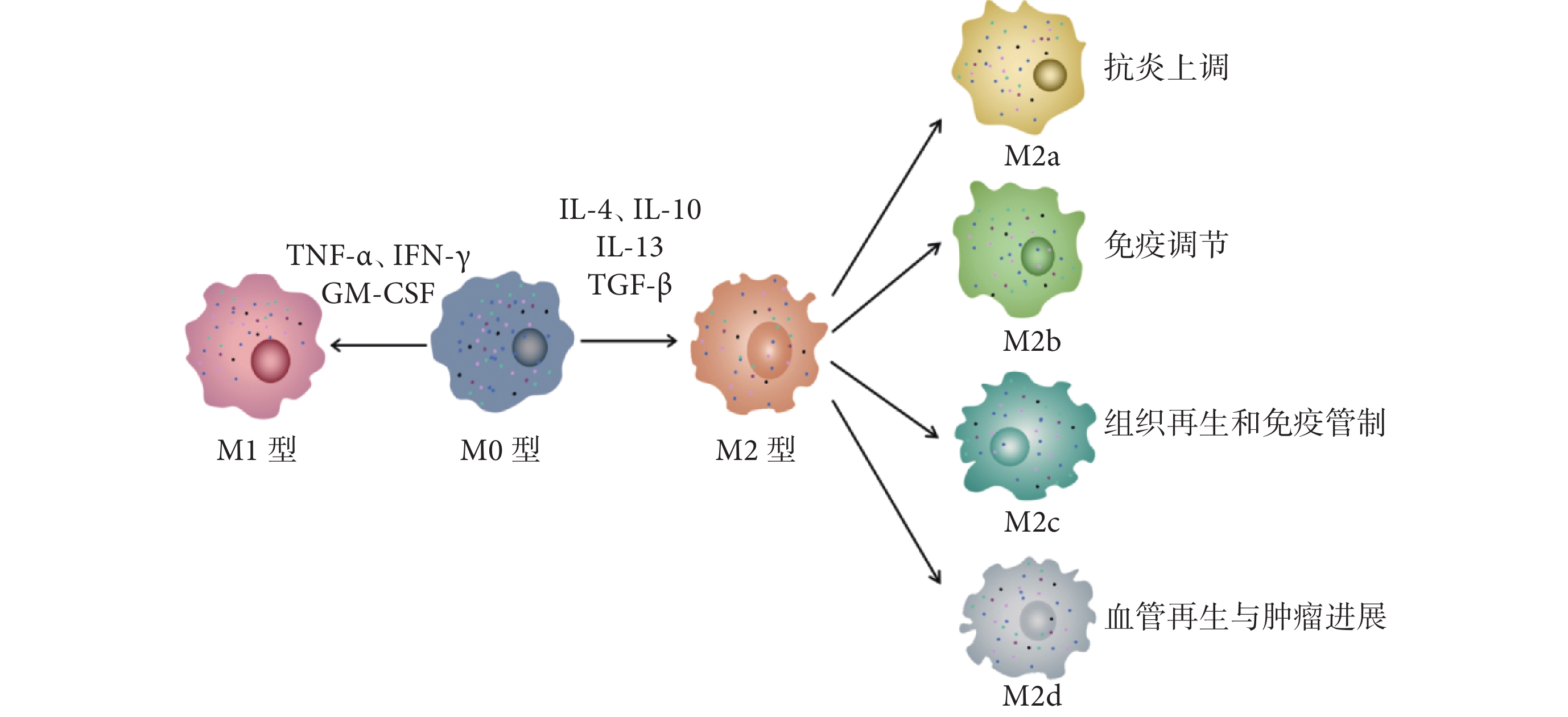
TAM是一種在TME中存在的巨噬細胞亞群。它們是由骨髓來源的單核細胞系分化而來,主要定位于腫瘤組織內。TAM根據其激活后的功能和效果可分為2個亞型:經典激活的M1型和選擇性激活的M2型TAM[7-8]。M1 型 TAM具有促炎性和抗腫瘤作用,能夠殺傷腫瘤細胞并促進免疫反應。此外,M1型通常是由病原體、脂多糖、粒細胞巨噬細胞集落刺激因子(granulocyte macrophage-colony stimulating factor,GM-CSF)、腫瘤壞死因子-α(tumor necrosis factor-α,TNF-α)和輔助型T細胞1(helper T cells type 1,Th1)細胞因子和干擾素-γ誘導激活[9]。M2型通常是由寄生蟲或真菌、免疫復合物、凋亡細胞、巨噬細胞集落刺激因子、白細胞介素-13、轉化生長因子β(transforming growth factor-β,TGF-β)和輔助型T細胞2(helper T cells type 2,Th2)激活的,具有抗炎性和促腫瘤作用,主要參與調節TME,促進腫瘤生長、轉移和血管生成[10]。TAM可以通過多種機制影響腫瘤的發展,包括促進腫瘤細胞增殖、抑制免疫細胞的活性、促進血管生成和調節TME[11]。因此,TAM在腫瘤的免疫治療中扮演著重要的角色,其活性和功能狀態對免疫治療的效果具有非常重要的影響。巨噬細胞極化過程見圖1。
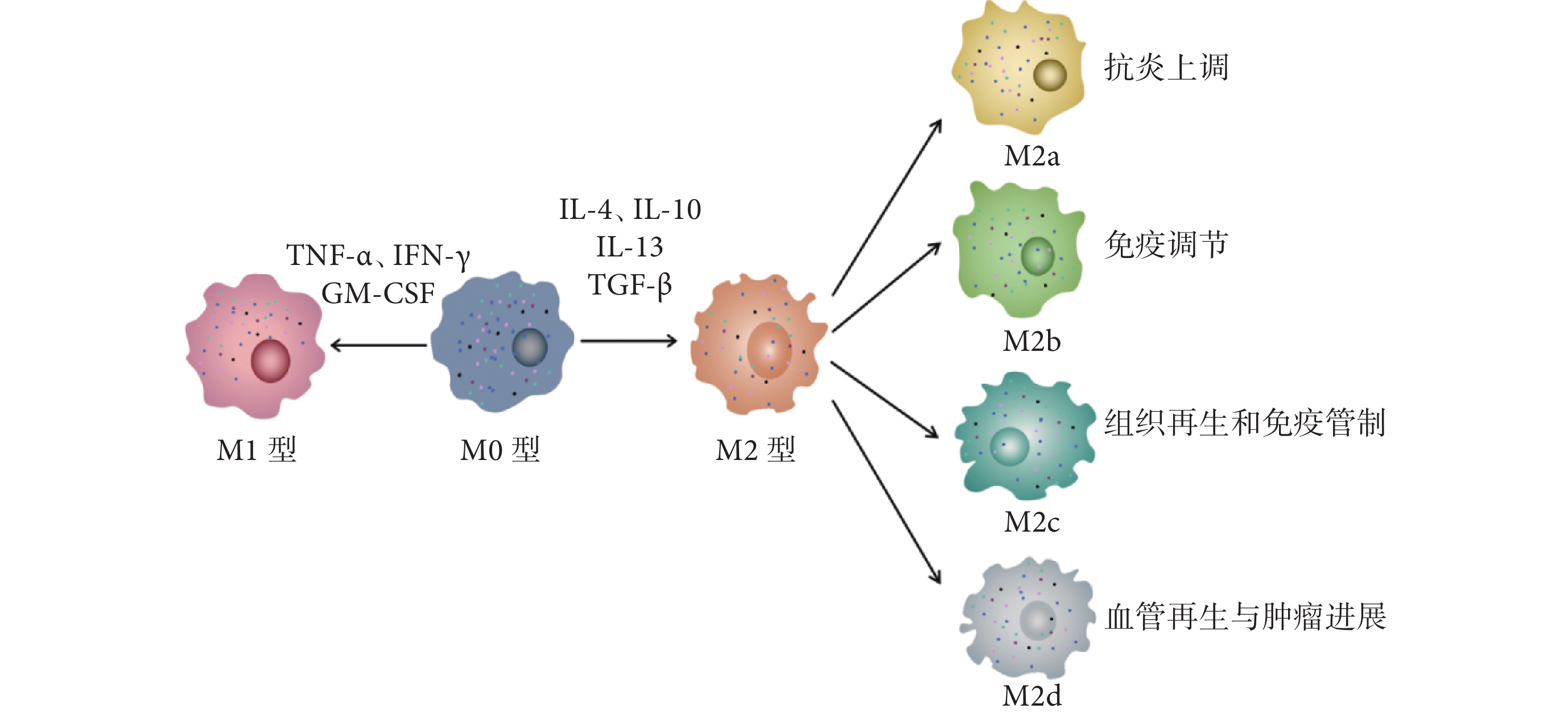 圖1
示巨噬細胞極化過程
圖1
示巨噬細胞極化過程
2 TAM參與介導胃癌的免疫耐藥
2.1 胃癌腫瘤免疫微環境(tumor immune microenvironment,TIME)的特殊性
胃癌TIME主要由腫瘤相關免疫細胞、免疫抑制性細胞、免疫激活細胞及腫瘤細胞本身共同組成。不同于其他腫瘤類型,胃癌的TIME具有一些獨特的特點。胃癌腫瘤中的淋巴細胞浸潤較為顯著,尤其是CD8+ 細胞(細胞毒性T細胞)[12]。不過,在一些胃癌患者中,免疫抑制環境較為明顯,導致這些淋巴細胞的功能受到抑制。CD8+ T細胞在胃癌免疫微環境中的作用至關重要,但其功能往往受到免疫檢查點分子如程序性死亡受體-1(programmed cell death protein-1,PD-1)/細胞程序性死亡配體-1(programmed cell death ligand-1,PD-L1)的抑制[13-14]。胃癌免疫微環境中常見的另一種免疫細胞類型是調節性T細胞。Tregs在胃癌微環境中的比例較高,這些細胞通過抑制抗腫瘤免疫反應,促進免疫逃逸和腫瘤進展[15]。此外,腫瘤相關中性粒細胞(tumor-associated neutrophils,TANs)也被認為在胃癌免疫微環境中發揮重要作用。研究發現,TANs不僅促進腫瘤的進展,還通過釋放促炎細胞因子增強免疫抑制環境[16]。在胃癌中,M2型巨噬細胞較為常見,且與腫瘤的免疫逃逸、轉移以及不良預后密切相關。幽門螺桿菌是胃癌的一個主要致病因子,它通過引發慢性胃炎、促進炎癥反應并改變免疫微環境,增加胃癌發生的風險。 幽門螺桿菌感染能通過誘導免疫系統產生慢性炎癥反應,改變胃癌的免疫微環境,推動腫瘤的進展[17]。胃癌的免疫微環境具有獨特性,表現為腫瘤細胞與免疫細胞之間復雜的相互作用。免疫逃逸機制、免疫抑制細胞的浸潤、微生物群落的影響以及代謝異常等共同塑造了胃癌的免疫微環境。這些特性使得胃癌的免疫治療充滿挑戰,因此,針對胃癌免疫微環境的精準調控和免疫治療策略的開發仍是當前研究的重點。
2.2 TAM參與胃癌免疫治療耐藥可能的機制
2.2.1 對T細胞活化和功能的影響
TAM可以抑制T細胞的活性,減弱它們對腫瘤細胞的識別和攻擊能力,導致腫瘤細胞逃避免疫監測,進而使腫瘤細胞對免疫治療產生耐藥性[18]。TGF-β已被證明通過抑制T細胞的活化和PD-L1的表達來影響抗PD-1/PD-L1免疫治療[19]。源自TAM的TGF-β通過磷酸化Smad2/3蛋白和抑制線粒體呼吸來降低T細胞中干擾素-γ(interferon gamma,IFN-γ)和顆粒酶B的表達水平,從而抑制T細胞活性[20]。TGF-β的表達與T細胞浸潤有關。TGF-β表達較高的腫瘤表現出較低的CD8+ T細胞浸潤。通過增加PD-L1的表達,前列腺素E2(prostaglandin E2,PGE2)可以抑制T細胞的活化和功能。作為環氧合酶2(Cyclooxygenase 2,COX-2)的下游,TME中PGE2的水平受COX-2和微粒體PGE2合酶1表達的調節[21]。T細胞活化的V結構域Ig抑制因子(V-domain Ig suppressor of T cell activation,VISTA)是一種由TAM表達的免疫檢查點配體,同時也是一種免疫抑制分子,它的表達可以減少T細胞增殖和細胞因子的產生,同時維持Tregs的功能[22]。VISTA的表達不僅與腫瘤細胞表面PD-L1的表達呈正相關,而且與患者的不良預后、病理分級和淋巴結狀態呈正相關。有研究[23]表明,在胃癌腫瘤組織中,唾液酸結合 Ig 樣凝集素 10(sialic-acid-binding Ig-like lectin 10,Siglec-10) 有助于調節 TAM 的免疫抑制特性,并促進 CD8+ T 細胞的耗竭。分泌磷蛋1(Secreted phosphoprotein 1,SPP1)陽性的TAMs與CD8+ T細胞之間的相互作用在mGC中發揮著重要的作用。通過生長分化因子15(growth differentiation factor 15,GDF15)轉化生長因子β受體2(transforming growth factor beta receptor 2,TGFBR2)信號通路,SPP1陽性TAMs能夠促進共抑制受體的表達,從而導致CD8+ T細胞的凋亡[24]。總體而言,TAM通過免疫抑制作用和調節免疫逃逸機制,顯著削弱T細胞的抗腫瘤功能,推動腫瘤的免疫逃逸進程。
2.2.2 形成抑制性TIME
TIME是TME的免疫環境亞類,主要由細胞毒性T細胞(cytotoxic T cells,CTL)、自然殺傷細胞(natural killer cell,NK)、腫瘤相關成纖維細胞(cancer-associated fibroblasts,CAFs)、髓源性抑制細胞(myeloid-derived suppressor cells,MDSCs)、TAM等免疫細胞組成。TAM可以分泌多種免疫抑制因子,如白細胞介素10、吲哚胺 2,3-雙加氧酶、TGF-β等促進免疫抑制性細胞的增加,也能誘導Treg、CAFs、樹突狀細胞(dendritic cell,DC)、 MDSCs等細胞共同形成抑制性TIME[25]。在TIME中,Treg和TAM之間的相互作用是不可忽視的。據報道,M2 型 TAM通過合成IL-10和TGF-β促進Treg的分化,并通過產生C-C基序趨化因子配體(C-C motif chemokine ligand 22,CCL22)加速Treg的招募,從而促進免疫抑制性表型的形成[26]。研究[27]表明,IL-10 分泌量高的M2型 TAM浸潤的胃癌患者在接受輔助化療時,治療反應和生存率較差,主要表現為Treg增加和 CD8 T 細胞功能受損。總之,TAM通過促進Tregs的招募和功能激活,增強免疫抑制作用,從而抑制效應T細胞的抗腫瘤活性,促進腫瘤的免疫逃逸。
2.2.3 TAM促進免疫逃逸
TAM可以分泌多種生長因子、細胞因子和蛋白酶促進腫瘤的生長、侵襲和轉移,并抑制免疫細胞的活性,從而幫助腫瘤細胞逃避免疫系統的監測和攻擊[28-29]。此外,TAM還可以通過抑制NK細胞的功能,減少它們對腫瘤細胞的殺傷作用,從而促進免疫逃逸。在小鼠異種移植物模型中,普列克底物蛋白2(plecksubstrate protein 2,PLEK2)通過磷脂酰肌醇3激酶(phosphatidyl inositol 3 kinase,PI3K)/蛋白激酶B(protein kinase B,AKT)通路促進轉錄因子特化蛋白1(transcription factor specialized protein 1,Sp1) 磷酸化,上調膜 I 型基質金屬蛋白酶(membrane type I matrix metalloproteinases,MT1-MMP)的表達,促使人類主要組織相容性復合體I類鏈相關基因A(human major histocompatibility complex class I chain related gene A,MICA)脫落,導致 NK細胞無法有效地識別和攻擊胃癌細胞,從而讓胃癌細胞逃避免疫系統的攻擊[30]。因此,TAM在胃癌中的存在和活性會影響免疫系統對腫瘤的監測和清除,促進腫瘤的發展和轉移,是胃癌免疫逃逸的重要因素之一。
在TIME中,還存在多種免疫抑制性信號通路和免疫抑制性細胞,如PD-L1/PD-1信號通路、細胞毒T淋巴細胞相關抗原4(cytotoxic t lymphocyte-associated antigen 4,CTLA-4)信號通路等,它們可以抑制免疫細胞的活性,促進腫瘤的免疫逃逸。因此,針對TIME中的免疫抑制機制進行干預,可能是提高腫瘤治療效果的重要策略之一。
3 靶向TAM可延緩胃癌進展
3.1 增加M1型TAM的比例
不同類型TAM的比例可以影響胃癌的預后,增加M1的數量是改善胃癌患者結局轉歸的常見策略。 M1型TAM通常表現出抗腫瘤的特性,可以通過釋放促炎細胞因子(如IL-12、TNF-α)和誘導T細胞的活化來促進抗腫瘤免疫應答,從而抑制腫瘤生長和擴散。有研究[31]發現,攜帶miR-16-5p的M1型TAM衍生的外泌體,能夠通過刺激PD-L1激活T細胞免疫反應,對胃癌進程具有抑制作用。該研究強調了通過增加外泌體中的miR-16-5p,M1表型有望成為為一種基于細胞水平的胃癌療法。同時,M1型TAM可以改變TME中的氧化還原平衡、代謝狀態和細胞間通訊,從而影響腫瘤細胞的行為和生存[32]。 研究[33]發現,活性氧(reactive oxygen species,ROS)作為信號分子發揮作用,通過共濟失調-毛細血管擴張突變(ataxia-telangiectasia mutated,ATM)和細胞周期檢查點激酶2(checkpoint kinase 2,Chk2)調節 M1 型TAM極化。進一步研究證明 Chk2 能夠在 T95 和 T195 位點磷酸化丙酮酸激酶 M2(pyruvate kinase M2,PKM2),促進糖酵解并實現TAM發生 M1 極化。 此外,Chk2 激活增加了 p21 的 Chk2 依賴性表達,誘導細胞周期停滯以進行隨后的 M1 極化。最后,感染脂多糖(lipopolysaccharides,LPS)的 Chk2 缺陷小鼠的肺部炎癥和 M1 巨噬細胞計數顯著降低。這些結果表明,抑制ROS-Chk2軸可以防止TAM的過度活化,并且該途徑可以靶向開發炎癥相關疾病的新療法。另一項研究[34]發現,在M0向M1極化過程中雙分支N-乙酰葡萄糖胺(bisecting N-acetylglucosamine,Bisecting GlcNAc)顯著減少,受損的Bisecting GlcNAc能夠推動M0向M1的極化。同時,還發現半乳糖凝集素3 結合蛋白(galactin-3-binding protein,Lgals3bp)的水平升高可以通過激活NF-кB通路促進M1極化。相反,激活的NF-кB通路顯著抑制了單酰基甘油酰基轉移酶 3(monoacylglyceryl acyltransferase 3,MGAT3)的轉錄,導致宿主蛋白Lgals3bp上的Bisecting GlcNAc修飾水平降低。該研究突出了糖基化對TAM極化的影響,說明通過糖基修飾的改變成為影響TAM的極化的一種方式。
綜上所述,增加M1型TAM的比例可能通過促進抗腫瘤免疫、促進腫瘤細胞凋亡、抑制腫瘤血管生成、調節TME等多種機制來改善胃癌的預后。
3.2 降低M2型TAM的比例
M2激活狀態存在異質性且功能不同,目前普遍根據刺激源以及轉錄變化將M2型巨噬細胞分成M2a、M2b、M2c和M2d 4個亞型[35-36]。目前可以通過直接或間接減少M2 表型的數量,作為抗腫瘤藥物的傳遞工具和M2重極化為M1表型3種方式降低TME中M2 型TAM的比例[37]。
3.2.1 減少M2表型的數量
一項研究[38]發現,環狀RNAATP8A1(circRNA ATP8A1,circATP8A1)能夠通過信號轉導和轉錄激活因子(signal transduction and activator of transcription 6,STAT6)途徑誘導巨噬細胞的 M2 極化,敲低circATP8A1 可在體外和體內顯著抑制胃癌的增殖和侵襲。總的來說,在胃癌細胞中,外泌體來源的 circATP8A1 能夠通過 circATP8A1/miR-1-3p/STAT6 軸誘導TAM發生 M2 極化,并誘導腫瘤進展。該結果表明, circATP8A1 能夠作為胃癌的潛在預后生物標志物和治療靶點。還有研究[39]發現,外泌體來源的MIR4435-2宿主基因(MIR4435-2 host gene,MIR4435-2HG)與胃癌患者的存活率呈負相關,抑制MIR4435-2HG的活性能夠降低胃癌細胞的活力和遷移能力(P<0.001)。通過人胃癌MKN45細胞來源的外泌體遞送MIR4435-2HG shRNA顯著逆轉了MKN45外泌體誘導的M2極化。該研究結果提示,MKN45細胞來源的外泌體遞送lncRNA MIR4435-2HG,能夠通過誘導TAM 向M2極化,從而促進胃癌發生。進一步研究[40]發現,敲除MIR4435-2HG可抑制胃癌細胞的增殖、侵襲、遷移和上皮-間充質轉化(epithelial-mesenchymal transition,EMT),加速胃癌細胞凋亡(P<0.01)。此外,MIR4435-2HG的缺失能夠減弱體內腫瘤的生長。靶向 miR-138-5p/Sox4 軸促進胃癌細胞的生長、轉移和 EMT。以上結果表明,MIR4435-2HG可能作為胃癌的潛在治療靶點。
3.2.2 抗腫瘤藥物的傳遞介質
TAM表現出顯著的歸巢特性,能夠通過特定信號在腫瘤組織中定向遷移和積聚。基于此特性,使通過TAM呈遞抗腫瘤藥物成為可能。 Au納米殼是具有二氧化硅核心和薄Au殼的納米顆粒。研究[41]發現,Au納米殼在與TAM共培養后,Au納米殼被吞噬、內化到TAM內部,這些TAM將攜帶的Au納米殼積累在腫瘤的缺氧核心區域,改善了Au納米粒子的靶向性并增強了腫瘤熱消融療法的療效。
3.2.3 加速M2型重極化為M1型
TAM表現出顯著的可塑性,能夠在各種因素的影響下進行表型轉換。 因此,TAM從M2到M1型的靶向重編程代表了腫瘤學研究的一個關鍵領域。 一項研究[42]開發了一種用甘露糖-聚乙二醇(mannose-polyethylene glycol,Man-PEG)修飾的多功能脂質體藥物遞送系統(DOX/R837@ManL)。該系統能夠有效調節TME的免疫抑制,并通過重極化M2 型TAM和誘導細胞死亡免疫原釋放促進癌癥免疫治療。有研究[43]表明,通過雙重靶向工程化的牛奶外泌體(milk exosome,mEoxs),成功將PDL-1特異性小干擾RNA(PDL-1-specific small interfering RNA,siPDL-1)有效地遞送到 M2 型TAM 中,將其轉化為 M1 型TAM,從而恢復 CD8+ T 細胞的免疫活性并改變TME。該研究結果顯示,在實驗中全身給藥 7D12-mExo-M2pep-siPDL1 表現出顯著的抗腫瘤活性,對原位表皮生長因子受體(epidermal growth factor receptor,EGFR)癌癥小鼠模型中的腫瘤生長有近 90% 的抑制作用。
3.3 嵌合抗原受體-巨噬細胞(chimeric antigen receptor macrophage therapy,CAR-T)療法
有研究[44-45]使用基因工程單核細胞和TAM作為抗腫瘤的治療方法,并取名為CAR-T,該療法的主要原理是通過基因工程技術,利用巨噬細胞的特性使其具備特定的抗腫瘤能力。一項研究[46]報道了人嵌合抗原受體巨噬細胞(chimeric antigen receptor macrophage,CAR-M)的初步開發,該研究發現,基于 CD3ζ 的 CAR 在人巨噬細胞中具有高度活性,能夠在不添加任何可溶性調理因子的情況下以 Syk 依賴性方式驅動吞噬作用并殺死攜帶靶標的腫瘤細胞。 為了建立人CAR-M細胞治療的轉化方法,另有研究[47]發現,在異種移植模型中,與用對照巨噬細胞治療的小鼠相比,CAR-M 治療顯著降低了腫瘤負荷,并延長了總生存期。還有研究[48]顯示,順鉑作為化療藥物通過Notch信號通路增加了CD133陽性(胃癌干細胞生物標志物)細胞;在BGC-823細胞中,CD133 CAR-T細胞促進了激活標志物的表達,包括CD27和CD69,并增強了T細胞活性的細胞因子分泌;同樣,CD133 CAR-T細胞和順鉑在KATO Ⅲ和MKN-28異種移植模型中顯著減緩了腫瘤進展。
4 結語
在TME中,TAM總體表現為促腫瘤生長,在胃癌的發生和發展過程中扮演著重要角色。因此,如何抑制TAM的促瘤作用將為未來的胃癌治療提供新的線索。然而,包括TAM的發生機制以及驅動胃癌TME中TAM表型變化的關鍵因素仍待進一步研究。 最近的臨床前和臨床研究顯示,針對TAM的治療在治療胃癌方面取得了令人滿意的效果。
重要聲明
利益沖突聲明:本文全體作者閱讀并理解了《中國普外基礎與臨床雜志》的政策聲明,我們沒有相互競爭的利益。
作者貢獻聲明:劉濤負責查閱文獻、起草和文章撰寫;余春燕和李桐桐負責大綱梳理和修訂文章結構;黃翌楚負責修訂論文格式;姜雷對文章重要論點給予指導性意見并對最終文稿內容進行審閱。
胃癌是高度惡性腫瘤,其死亡率居全球惡性腫瘤第5位[1]。我國胃癌發病率較高,發病率及死亡率均居惡性腫瘤的第3位[2]。近年來,在低風險和高風險國家,年輕人的胃癌發病率在逐步上升[3]。化療是轉移性胃癌(metastatic gastric cancer,mGC)的主要治療方法,對于接受常規化療的患者,中位總生存期(overall survival,OS)約為12個月[4]。免疫療法在胃癌治療方面的效果顯著,已成為晚期胃癌的一線治療方案[5]。免疫檢查點抑制劑(immune checkpoint inhibitors,ICIs)等新型免疫治療的出現在一定程度上改變了胃癌患者的治療。然而,只有少數晚期胃癌患者對ICIs治療有持久反應,且ICIs的療效非常有限[6]。腫瘤相關巨噬細胞(tumor-associated macrophage,TAM)在胃癌的免疫治療中發揮重要作用的同時,可能會導致免疫治療的耐藥性。這種耐藥性可能是由于TAM的免疫抑制作用,或者是由于它們在腫瘤微環境(tumor microenvironment,TME)中的功能受到干擾。因此,了解TAM在免疫治療中的作用和調控機制對于克服免疫治療耐藥性具有重要意義。筆者現就TAM的發生發展及其與癌癥的關系予以綜述,重點闡述TAM在胃癌免疫治療及耐藥方面可能存在的機制,以及針對耐藥可能的治療手段。
1 TAM的起源和分類
TAM是一種在TME中存在的巨噬細胞亞群。它們是由骨髓來源的單核細胞系分化而來,主要定位于腫瘤組織內。TAM根據其激活后的功能和效果可分為2個亞型:經典激活的M1型和選擇性激活的M2型TAM[7-8]。M1 型 TAM具有促炎性和抗腫瘤作用,能夠殺傷腫瘤細胞并促進免疫反應。此外,M1型通常是由病原體、脂多糖、粒細胞巨噬細胞集落刺激因子(granulocyte macrophage-colony stimulating factor,GM-CSF)、腫瘤壞死因子-α(tumor necrosis factor-α,TNF-α)和輔助型T細胞1(helper T cells type 1,Th1)細胞因子和干擾素-γ誘導激活[9]。M2型通常是由寄生蟲或真菌、免疫復合物、凋亡細胞、巨噬細胞集落刺激因子、白細胞介素-13、轉化生長因子β(transforming growth factor-β,TGF-β)和輔助型T細胞2(helper T cells type 2,Th2)激活的,具有抗炎性和促腫瘤作用,主要參與調節TME,促進腫瘤生長、轉移和血管生成[10]。TAM可以通過多種機制影響腫瘤的發展,包括促進腫瘤細胞增殖、抑制免疫細胞的活性、促進血管生成和調節TME[11]。因此,TAM在腫瘤的免疫治療中扮演著重要的角色,其活性和功能狀態對免疫治療的效果具有非常重要的影響。巨噬細胞極化過程見圖1。
圖1
示巨噬細胞極化過程
2 TAM參與介導胃癌的免疫耐藥
2.1 胃癌腫瘤免疫微環境(tumor immune microenvironment,TIME)的特殊性
胃癌TIME主要由腫瘤相關免疫細胞、免疫抑制性細胞、免疫激活細胞及腫瘤細胞本身共同組成。不同于其他腫瘤類型,胃癌的TIME具有一些獨特的特點。胃癌腫瘤中的淋巴細胞浸潤較為顯著,尤其是CD8+ 細胞(細胞毒性T細胞)[12]。不過,在一些胃癌患者中,免疫抑制環境較為明顯,導致這些淋巴細胞的功能受到抑制。CD8+ T細胞在胃癌免疫微環境中的作用至關重要,但其功能往往受到免疫檢查點分子如程序性死亡受體-1(programmed cell death protein-1,PD-1)/細胞程序性死亡配體-1(programmed cell death ligand-1,PD-L1)的抑制[13-14]。胃癌免疫微環境中常見的另一種免疫細胞類型是調節性T細胞。Tregs在胃癌微環境中的比例較高,這些細胞通過抑制抗腫瘤免疫反應,促進免疫逃逸和腫瘤進展[15]。此外,腫瘤相關中性粒細胞(tumor-associated neutrophils,TANs)也被認為在胃癌免疫微環境中發揮重要作用。研究發現,TANs不僅促進腫瘤的進展,還通過釋放促炎細胞因子增強免疫抑制環境[16]。在胃癌中,M2型巨噬細胞較為常見,且與腫瘤的免疫逃逸、轉移以及不良預后密切相關。幽門螺桿菌是胃癌的一個主要致病因子,它通過引發慢性胃炎、促進炎癥反應并改變免疫微環境,增加胃癌發生的風險。 幽門螺桿菌感染能通過誘導免疫系統產生慢性炎癥反應,改變胃癌的免疫微環境,推動腫瘤的進展[17]。胃癌的免疫微環境具有獨特性,表現為腫瘤細胞與免疫細胞之間復雜的相互作用。免疫逃逸機制、免疫抑制細胞的浸潤、微生物群落的影響以及代謝異常等共同塑造了胃癌的免疫微環境。這些特性使得胃癌的免疫治療充滿挑戰,因此,針對胃癌免疫微環境的精準調控和免疫治療策略的開發仍是當前研究的重點。
2.2 TAM參與胃癌免疫治療耐藥可能的機制
2.2.1 對T細胞活化和功能的影響
TAM可以抑制T細胞的活性,減弱它們對腫瘤細胞的識別和攻擊能力,導致腫瘤細胞逃避免疫監測,進而使腫瘤細胞對免疫治療產生耐藥性[18]。TGF-β已被證明通過抑制T細胞的活化和PD-L1的表達來影響抗PD-1/PD-L1免疫治療[19]。源自TAM的TGF-β通過磷酸化Smad2/3蛋白和抑制線粒體呼吸來降低T細胞中干擾素-γ(interferon gamma,IFN-γ)和顆粒酶B的表達水平,從而抑制T細胞活性[20]。TGF-β的表達與T細胞浸潤有關。TGF-β表達較高的腫瘤表現出較低的CD8+ T細胞浸潤。通過增加PD-L1的表達,前列腺素E2(prostaglandin E2,PGE2)可以抑制T細胞的活化和功能。作為環氧合酶2(Cyclooxygenase 2,COX-2)的下游,TME中PGE2的水平受COX-2和微粒體PGE2合酶1表達的調節[21]。T細胞活化的V結構域Ig抑制因子(V-domain Ig suppressor of T cell activation,VISTA)是一種由TAM表達的免疫檢查點配體,同時也是一種免疫抑制分子,它的表達可以減少T細胞增殖和細胞因子的產生,同時維持Tregs的功能[22]。VISTA的表達不僅與腫瘤細胞表面PD-L1的表達呈正相關,而且與患者的不良預后、病理分級和淋巴結狀態呈正相關。有研究[23]表明,在胃癌腫瘤組織中,唾液酸結合 Ig 樣凝集素 10(sialic-acid-binding Ig-like lectin 10,Siglec-10) 有助于調節 TAM 的免疫抑制特性,并促進 CD8+ T 細胞的耗竭。分泌磷蛋1(Secreted phosphoprotein 1,SPP1)陽性的TAMs與CD8+ T細胞之間的相互作用在mGC中發揮著重要的作用。通過生長分化因子15(growth differentiation factor 15,GDF15)轉化生長因子β受體2(transforming growth factor beta receptor 2,TGFBR2)信號通路,SPP1陽性TAMs能夠促進共抑制受體的表達,從而導致CD8+ T細胞的凋亡[24]。總體而言,TAM通過免疫抑制作用和調節免疫逃逸機制,顯著削弱T細胞的抗腫瘤功能,推動腫瘤的免疫逃逸進程。
2.2.2 形成抑制性TIME
TIME是TME的免疫環境亞類,主要由細胞毒性T細胞(cytotoxic T cells,CTL)、自然殺傷細胞(natural killer cell,NK)、腫瘤相關成纖維細胞(cancer-associated fibroblasts,CAFs)、髓源性抑制細胞(myeloid-derived suppressor cells,MDSCs)、TAM等免疫細胞組成。TAM可以分泌多種免疫抑制因子,如白細胞介素10、吲哚胺 2,3-雙加氧酶、TGF-β等促進免疫抑制性細胞的增加,也能誘導Treg、CAFs、樹突狀細胞(dendritic cell,DC)、 MDSCs等細胞共同形成抑制性TIME[25]。在TIME中,Treg和TAM之間的相互作用是不可忽視的。據報道,M2 型 TAM通過合成IL-10和TGF-β促進Treg的分化,并通過產生C-C基序趨化因子配體(C-C motif chemokine ligand 22,CCL22)加速Treg的招募,從而促進免疫抑制性表型的形成[26]。研究[27]表明,IL-10 分泌量高的M2型 TAM浸潤的胃癌患者在接受輔助化療時,治療反應和生存率較差,主要表現為Treg增加和 CD8 T 細胞功能受損。總之,TAM通過促進Tregs的招募和功能激活,增強免疫抑制作用,從而抑制效應T細胞的抗腫瘤活性,促進腫瘤的免疫逃逸。
2.2.3 TAM促進免疫逃逸
TAM可以分泌多種生長因子、細胞因子和蛋白酶促進腫瘤的生長、侵襲和轉移,并抑制免疫細胞的活性,從而幫助腫瘤細胞逃避免疫系統的監測和攻擊[28-29]。此外,TAM還可以通過抑制NK細胞的功能,減少它們對腫瘤細胞的殺傷作用,從而促進免疫逃逸。在小鼠異種移植物模型中,普列克底物蛋白2(plecksubstrate protein 2,PLEK2)通過磷脂酰肌醇3激酶(phosphatidyl inositol 3 kinase,PI3K)/蛋白激酶B(protein kinase B,AKT)通路促進轉錄因子特化蛋白1(transcription factor specialized protein 1,Sp1) 磷酸化,上調膜 I 型基質金屬蛋白酶(membrane type I matrix metalloproteinases,MT1-MMP)的表達,促使人類主要組織相容性復合體I類鏈相關基因A(human major histocompatibility complex class I chain related gene A,MICA)脫落,導致 NK細胞無法有效地識別和攻擊胃癌細胞,從而讓胃癌細胞逃避免疫系統的攻擊[30]。因此,TAM在胃癌中的存在和活性會影響免疫系統對腫瘤的監測和清除,促進腫瘤的發展和轉移,是胃癌免疫逃逸的重要因素之一。
在TIME中,還存在多種免疫抑制性信號通路和免疫抑制性細胞,如PD-L1/PD-1信號通路、細胞毒T淋巴細胞相關抗原4(cytotoxic t lymphocyte-associated antigen 4,CTLA-4)信號通路等,它們可以抑制免疫細胞的活性,促進腫瘤的免疫逃逸。因此,針對TIME中的免疫抑制機制進行干預,可能是提高腫瘤治療效果的重要策略之一。
3 靶向TAM可延緩胃癌進展
3.1 增加M1型TAM的比例
不同類型TAM的比例可以影響胃癌的預后,增加M1的數量是改善胃癌患者結局轉歸的常見策略。 M1型TAM通常表現出抗腫瘤的特性,可以通過釋放促炎細胞因子(如IL-12、TNF-α)和誘導T細胞的活化來促進抗腫瘤免疫應答,從而抑制腫瘤生長和擴散。有研究[31]發現,攜帶miR-16-5p的M1型TAM衍生的外泌體,能夠通過刺激PD-L1激活T細胞免疫反應,對胃癌進程具有抑制作用。該研究強調了通過增加外泌體中的miR-16-5p,M1表型有望成為為一種基于細胞水平的胃癌療法。同時,M1型TAM可以改變TME中的氧化還原平衡、代謝狀態和細胞間通訊,從而影響腫瘤細胞的行為和生存[32]。 研究[33]發現,活性氧(reactive oxygen species,ROS)作為信號分子發揮作用,通過共濟失調-毛細血管擴張突變(ataxia-telangiectasia mutated,ATM)和細胞周期檢查點激酶2(checkpoint kinase 2,Chk2)調節 M1 型TAM極化。進一步研究證明 Chk2 能夠在 T95 和 T195 位點磷酸化丙酮酸激酶 M2(pyruvate kinase M2,PKM2),促進糖酵解并實現TAM發生 M1 極化。 此外,Chk2 激活增加了 p21 的 Chk2 依賴性表達,誘導細胞周期停滯以進行隨后的 M1 極化。最后,感染脂多糖(lipopolysaccharides,LPS)的 Chk2 缺陷小鼠的肺部炎癥和 M1 巨噬細胞計數顯著降低。這些結果表明,抑制ROS-Chk2軸可以防止TAM的過度活化,并且該途徑可以靶向開發炎癥相關疾病的新療法。另一項研究[34]發現,在M0向M1極化過程中雙分支N-乙酰葡萄糖胺(bisecting N-acetylglucosamine,Bisecting GlcNAc)顯著減少,受損的Bisecting GlcNAc能夠推動M0向M1的極化。同時,還發現半乳糖凝集素3 結合蛋白(galactin-3-binding protein,Lgals3bp)的水平升高可以通過激活NF-кB通路促進M1極化。相反,激活的NF-кB通路顯著抑制了單酰基甘油酰基轉移酶 3(monoacylglyceryl acyltransferase 3,MGAT3)的轉錄,導致宿主蛋白Lgals3bp上的Bisecting GlcNAc修飾水平降低。該研究突出了糖基化對TAM極化的影響,說明通過糖基修飾的改變成為影響TAM的極化的一種方式。
綜上所述,增加M1型TAM的比例可能通過促進抗腫瘤免疫、促進腫瘤細胞凋亡、抑制腫瘤血管生成、調節TME等多種機制來改善胃癌的預后。
3.2 降低M2型TAM的比例
M2激活狀態存在異質性且功能不同,目前普遍根據刺激源以及轉錄變化將M2型巨噬細胞分成M2a、M2b、M2c和M2d 4個亞型[35-36]。目前可以通過直接或間接減少M2 表型的數量,作為抗腫瘤藥物的傳遞工具和M2重極化為M1表型3種方式降低TME中M2 型TAM的比例[37]。
3.2.1 減少M2表型的數量
一項研究[38]發現,環狀RNAATP8A1(circRNA ATP8A1,circATP8A1)能夠通過信號轉導和轉錄激活因子(signal transduction and activator of transcription 6,STAT6)途徑誘導巨噬細胞的 M2 極化,敲低circATP8A1 可在體外和體內顯著抑制胃癌的增殖和侵襲。總的來說,在胃癌細胞中,外泌體來源的 circATP8A1 能夠通過 circATP8A1/miR-1-3p/STAT6 軸誘導TAM發生 M2 極化,并誘導腫瘤進展。該結果表明, circATP8A1 能夠作為胃癌的潛在預后生物標志物和治療靶點。還有研究[39]發現,外泌體來源的MIR4435-2宿主基因(MIR4435-2 host gene,MIR4435-2HG)與胃癌患者的存活率呈負相關,抑制MIR4435-2HG的活性能夠降低胃癌細胞的活力和遷移能力(P<0.001)。通過人胃癌MKN45細胞來源的外泌體遞送MIR4435-2HG shRNA顯著逆轉了MKN45外泌體誘導的M2極化。該研究結果提示,MKN45細胞來源的外泌體遞送lncRNA MIR4435-2HG,能夠通過誘導TAM 向M2極化,從而促進胃癌發生。進一步研究[40]發現,敲除MIR4435-2HG可抑制胃癌細胞的增殖、侵襲、遷移和上皮-間充質轉化(epithelial-mesenchymal transition,EMT),加速胃癌細胞凋亡(P<0.01)。此外,MIR4435-2HG的缺失能夠減弱體內腫瘤的生長。靶向 miR-138-5p/Sox4 軸促進胃癌細胞的生長、轉移和 EMT。以上結果表明,MIR4435-2HG可能作為胃癌的潛在治療靶點。
3.2.2 抗腫瘤藥物的傳遞介質
TAM表現出顯著的歸巢特性,能夠通過特定信號在腫瘤組織中定向遷移和積聚。基于此特性,使通過TAM呈遞抗腫瘤藥物成為可能。 Au納米殼是具有二氧化硅核心和薄Au殼的納米顆粒。研究[41]發現,Au納米殼在與TAM共培養后,Au納米殼被吞噬、內化到TAM內部,這些TAM將攜帶的Au納米殼積累在腫瘤的缺氧核心區域,改善了Au納米粒子的靶向性并增強了腫瘤熱消融療法的療效。
3.2.3 加速M2型重極化為M1型
TAM表現出顯著的可塑性,能夠在各種因素的影響下進行表型轉換。 因此,TAM從M2到M1型的靶向重編程代表了腫瘤學研究的一個關鍵領域。 一項研究[42]開發了一種用甘露糖-聚乙二醇(mannose-polyethylene glycol,Man-PEG)修飾的多功能脂質體藥物遞送系統(DOX/R837@ManL)。該系統能夠有效調節TME的免疫抑制,并通過重極化M2 型TAM和誘導細胞死亡免疫原釋放促進癌癥免疫治療。有研究[43]表明,通過雙重靶向工程化的牛奶外泌體(milk exosome,mEoxs),成功將PDL-1特異性小干擾RNA(PDL-1-specific small interfering RNA,siPDL-1)有效地遞送到 M2 型TAM 中,將其轉化為 M1 型TAM,從而恢復 CD8+ T 細胞的免疫活性并改變TME。該研究結果顯示,在實驗中全身給藥 7D12-mExo-M2pep-siPDL1 表現出顯著的抗腫瘤活性,對原位表皮生長因子受體(epidermal growth factor receptor,EGFR)癌癥小鼠模型中的腫瘤生長有近 90% 的抑制作用。
3.3 嵌合抗原受體-巨噬細胞(chimeric antigen receptor macrophage therapy,CAR-T)療法
有研究[44-45]使用基因工程單核細胞和TAM作為抗腫瘤的治療方法,并取名為CAR-T,該療法的主要原理是通過基因工程技術,利用巨噬細胞的特性使其具備特定的抗腫瘤能力。一項研究[46]報道了人嵌合抗原受體巨噬細胞(chimeric antigen receptor macrophage,CAR-M)的初步開發,該研究發現,基于 CD3ζ 的 CAR 在人巨噬細胞中具有高度活性,能夠在不添加任何可溶性調理因子的情況下以 Syk 依賴性方式驅動吞噬作用并殺死攜帶靶標的腫瘤細胞。 為了建立人CAR-M細胞治療的轉化方法,另有研究[47]發現,在異種移植模型中,與用對照巨噬細胞治療的小鼠相比,CAR-M 治療顯著降低了腫瘤負荷,并延長了總生存期。還有研究[48]顯示,順鉑作為化療藥物通過Notch信號通路增加了CD133陽性(胃癌干細胞生物標志物)細胞;在BGC-823細胞中,CD133 CAR-T細胞促進了激活標志物的表達,包括CD27和CD69,并增強了T細胞活性的細胞因子分泌;同樣,CD133 CAR-T細胞和順鉑在KATO Ⅲ和MKN-28異種移植模型中顯著減緩了腫瘤進展。
4 結語
在TME中,TAM總體表現為促腫瘤生長,在胃癌的發生和發展過程中扮演著重要角色。因此,如何抑制TAM的促瘤作用將為未來的胃癌治療提供新的線索。然而,包括TAM的發生機制以及驅動胃癌TME中TAM表型變化的關鍵因素仍待進一步研究。 最近的臨床前和臨床研究顯示,針對TAM的治療在治療胃癌方面取得了令人滿意的效果。
重要聲明
利益沖突聲明:本文全體作者閱讀并理解了《中國普外基礎與臨床雜志》的政策聲明,我們沒有相互競爭的利益。
作者貢獻聲明:劉濤負責查閱文獻、起草和文章撰寫;余春燕和李桐桐負責大綱梳理和修訂文章結構;黃翌楚負責修訂論文格式;姜雷對文章重要論點給予指導性意見并對最終文稿內容進行審閱。