

隆突性皮膚纖維肉瘤(dermatofibrosarcoma protuberans,DFSP)是一種發生于淺表的局部侵襲性纖維母細胞腫瘤,具有標志性COL1A1::PDGFB融合基因或其他相關融合[1]。經典型DFSP為低度惡性腫瘤,有10%~16%的經典型DFSP可通過高級別轉化形成纖維肉瘤型DFSP(fibrosarcomatous DFSP,FS-DFSP),生物學行為表現為侵襲性增強,具備遠處轉移潛能[2]。該腫瘤最常見的轉移部位為肺,還可見轉移至腦、骨等,單一腹腔轉移性FS-DFSP非常罕見。本研究報道了四川大學華西醫院(簡稱“我院”)于2023年診斷的1例腹腔轉移性FS-DFSP,該病例在診斷上極具挑戰性,現將其臨床病理資料及診治經過進行回顧,并結合國內外文獻報道進行分析,以期提高對疑難病例的認識、診斷及鑒別診斷水平。
1 臨床資料
1.1 一般資料
患者,女,51歲,因“反復左下腹痛6個月”于當地醫院就診。10年前因左側會陰部包塊行手術切除,外院診斷為“皮下纖維性腫瘤”,未能進一步分型。
1.2 外院診斷及處理
查體:左髖部捫及包塊,觸診大小約14 cm×10 cm×8 cm,皮膚表面張力高,見靜脈充盈明顯,皮溫較對側高,包塊表面光滑、質韌、有壓痛、與周圍組織邊界不清、活動度差。輔助檢查:CA125 59.3 U/mL(正常參考值<35.0 U/mL),CA72-4 8.0 U/mL(正常參考值<6.9 U/mL)。影像學檢查:① 腹部增強MRI檢查示腹腔左側一類圓形巨大腫塊,大小約14.2 cm×11.7 cm,T1WI呈稍低信號及部分為高信號,T2WI呈高信號伴散在少許低信號,增強掃描實性部分呈持續性不均勻強化,病灶與脾門區結構分界不清,胰腺受壓后移,胃及左腎受壓變形、移位,考慮惡性腫瘤可能性大。② 骨盆平掃MRI檢查示左側髖部肌間隙內不規則腫塊影,大小約14.0 cm×11.0 cm×5.8 cm,T1WI以等信號為主,其內散在點狀高信號影,T2信號混雜,以等、稍高信號為主,邊界欠清晰,鄰近臀大肌稍腫脹,臀大肌、臀小肌受壓推擠,考慮惡性腫瘤可能性大。行超聲引導下腹腔包塊穿刺活檢,病理診斷考慮脂肪源性肉瘤。臨床首先行腹腔包塊切除,術中見:腹腔左側一巨大腫塊,大小約14.2 cm×12.0 cm×10.0 cm,包膜較完整,腫瘤周圍與胃、結腸、脾及胰體尾粘連。大體見灰褐色包塊1個,大小13.0 cm×12.0 cm×9.5 cm,包塊表面與胰腺粘連,似有包膜,切面灰白灰褐,質嫩,局部似有出血,呈囊性變,囊內含血凝塊樣物。當地醫院病理診斷:“腹腔梭形細胞惡性腫瘤,類型不能除外上皮樣血管內皮瘤或去分化脂肪肉瘤”。患者為進一步明確腫瘤類型,于2023年8月至我院進行病理會診。
腹腔手術1個月后患者行髖部包塊切除,當地醫院病理診斷:“髖部梭形細胞惡性腫瘤”。患者將髖部包塊病理切片送至我院進行病理會診。
1.3 我院病理會診情況
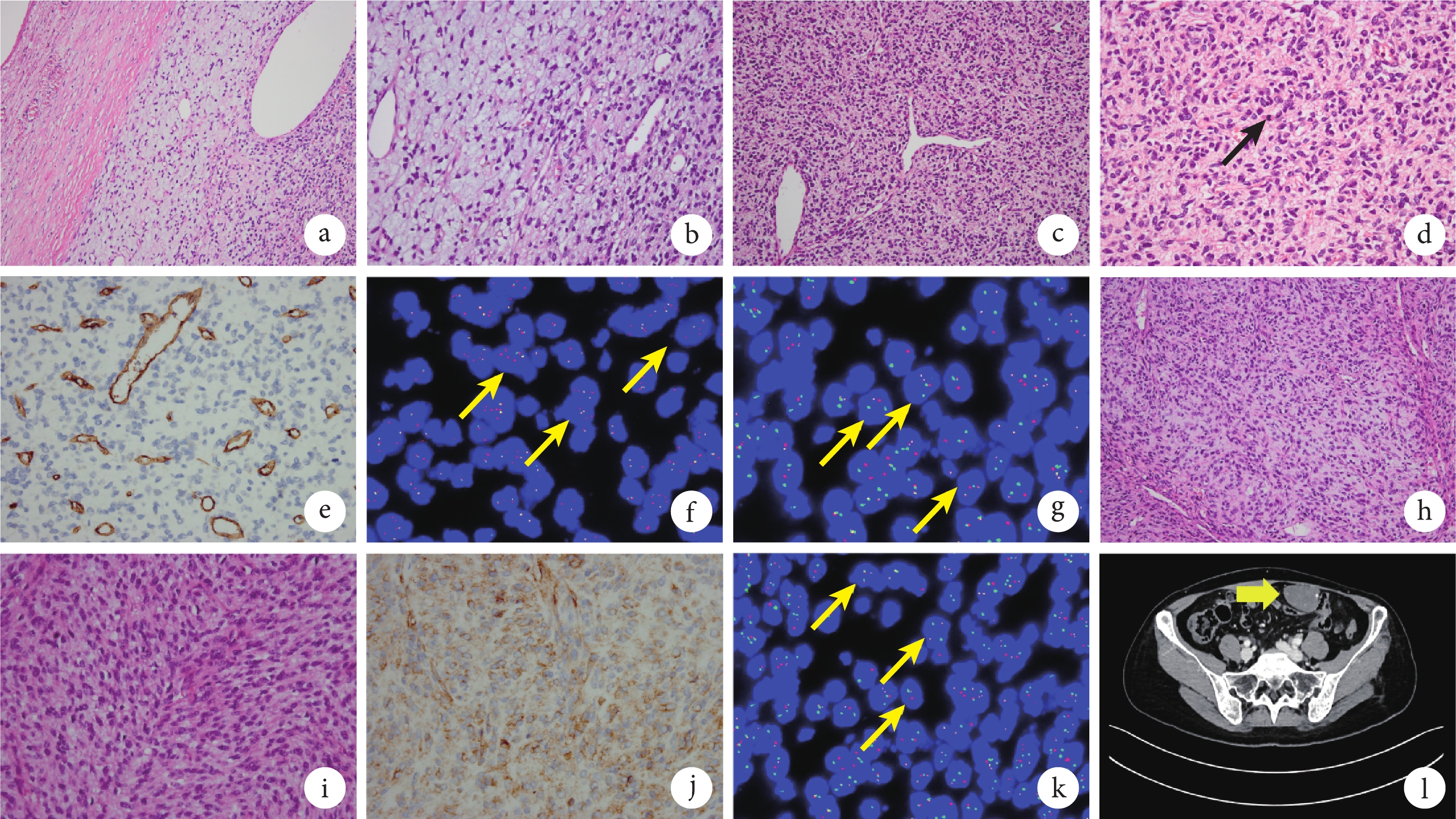
腹腔腫物鏡下可見腫瘤邊界清楚(圖1a),間質疏松,繼發黏液樣變(圖1b),其中含較多薄壁小血管,局灶可見血管外皮瘤樣構象(圖1c)。腫瘤主要由中度以上異型性的短梭形或卵圓形細胞組成,呈不規則排列(圖1d)。細胞核染色質呈粗顆粒狀,核仁不明顯,胞質呈嗜酸性。核分裂象易見(約10個/10 HPF),會診切片未見出血壞死。免疫組織化學結果顯示,腫瘤細胞CD34(圖1e)、結蛋白(Desmin)、S100、CD117和DOG1均為陰性,Ki?67陽性指數約為15%。熒光原位雜交(fluorescence in situ hybridization,FISH)檢測檢出PDGFB基因不平衡重排(圖1f)及COL1A1::PDGFB基因融合(圖1g),未檢出MDM2基因擴增、未檢出DDIT3基因重排。病理診斷為腹腔軟組織惡性腫瘤(肉瘤),結合免疫組織化學及基因檢測結果,亞型診斷為FS-DFSP;FNCLCC(法國國家癌癥中心聯盟)分級為2級。本病例首先需排除腹腔轉移性腫瘤,應積極查找原發病灶。
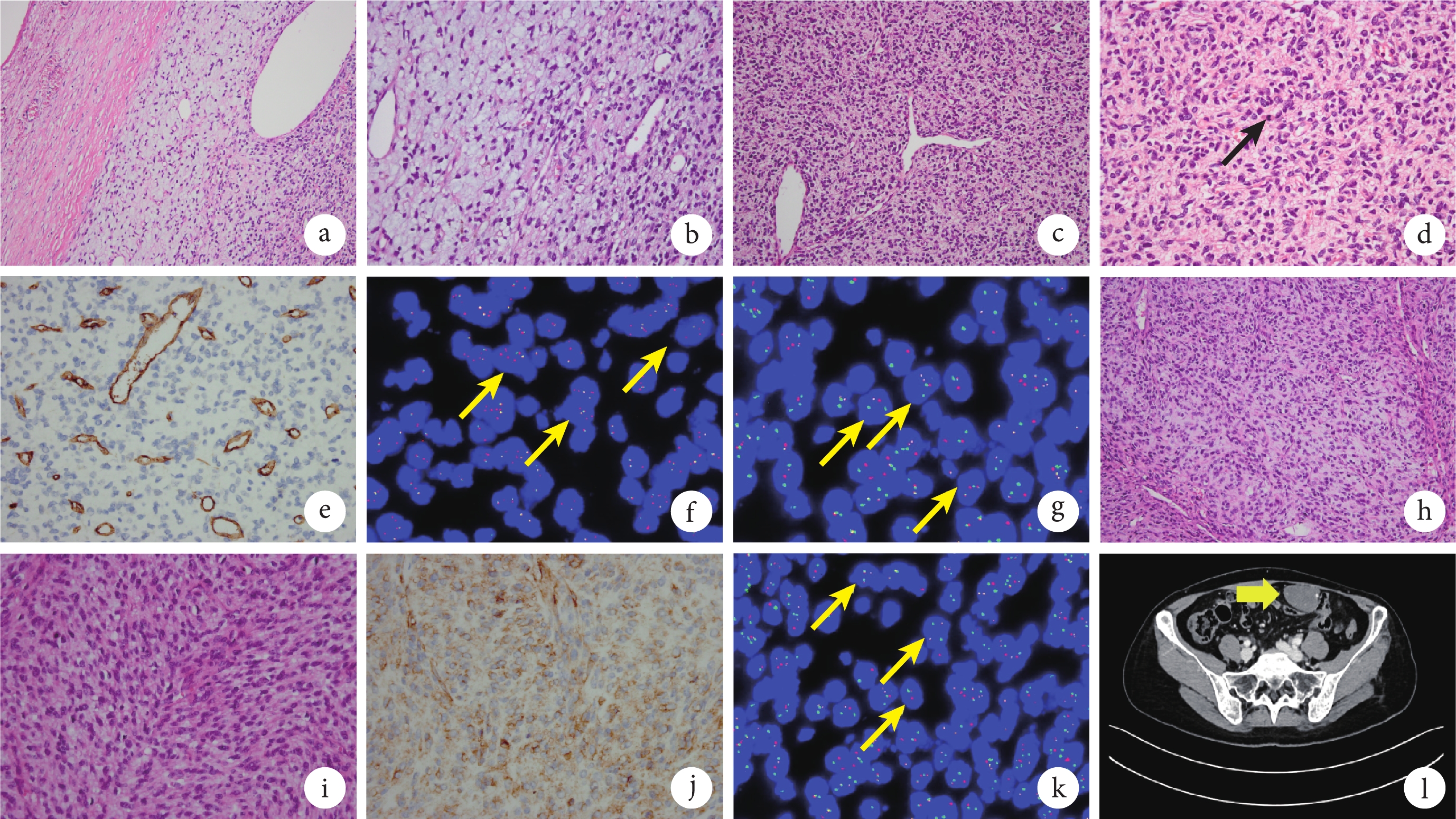 圖1
示該例患者的腹腔腫瘤和髖部腫瘤的組織學、免疫表型和分子遺傳學特征,以及明確FS-DFSP診斷后治療期間的CT復查結果
圖1
示該例患者的腹腔腫瘤和髖部腫瘤的組織學、免疫表型和分子遺傳學特征,以及明確FS-DFSP診斷后治療期間的CT復查結果
a~g為腹腔腫瘤結果;h~l為髖部腫瘤結果;a:腹腔腫瘤呈結節狀,邊界清楚(HE ×200);b:腫瘤間質疏松,繼發黏液樣變,間質內可見豐富的薄壁小血管(HE ×400);c:腫瘤間質部分區域可見血管外皮瘤樣構象(HE ×200);d:腫瘤由短梭形或卵圓形細胞組成,排列不規則,核分裂象活躍(黑箭,HE ×400);e:腹腔腫瘤細胞CD34呈陰性(EnVision法 ×400);f:FISH結果提示PDGFB基因重排(黃箭),PDGFB基因為紅色和綠色信號;g:FISH結果提示存在COL1A1::PDGFB融合基因(黃箭),COL1A1基因為紅色信號,PDGFB基因為綠色信號;h:髖部腫瘤大部分區域形態與腹腔腫瘤相似,由短梭形或卵圓形細胞呈不規則或短束狀結構排列組成(HE ×200);i:髖部腫瘤部分區域可見梭形細胞呈魚骨樣排列(HE ×200);j:髖部腫瘤CD34部分陽性(EnVision法 ×400);k:FISH結果提示存在COL1A1::PDGFB融合基因(黃箭),COL1A1基因為紅色信號,PDGFB基因為綠色信號;l:腹腔包塊切除術后15個月CT圖像示腹腔轉移灶復發,增強掃描明顯強化(黃箭)
髖部包塊鏡下觀察:髖部病變大部分區域形態與腹腔腫瘤相似(圖1h),部分區域可見中-重度異型性的梭形腫瘤細胞,呈束狀或魚骨樣結構排列(圖1i),核分裂象活躍(27個/10 HPF),部分區域可見壞死;腫瘤細胞CD34部分陽性(圖1j),其余標記與腹腔病變一致,信號轉導與轉錄激活因子6(signal transducer and activator of transcription-6,STAT6)和廣譜原肌球蛋白受體激酶(pan-tropomyosin receptor kinase,pan-TRK)均呈陰性,H3K27Me3未缺失;Ki?67陽性指數約為40%。FISH檢測檢出PDGFB基因不平衡重排及COL1A1::PDGFB基因融合(圖1k),未檢出CDKN2A基因缺失。病理診斷為髖部軟組織惡性腫瘤(肉瘤)。結合臨床、形態學、免疫組織化學及基因檢測結果,亞型診斷為FS-DFSP,支持髖部包塊與腹腔包塊為同源性腫瘤。
經臨床及病理軟組織專科多學科會診,聯系臨床,發現髖部腫瘤切除范圍大,與會陰部手術范圍相連。結合病理特征分析,考慮髖部腫瘤系會陰部腫瘤廣泛浸潤累及所致,腹腔腫瘤為轉移性FS-DFSP。
1.4 治療及隨訪
患者髖部包塊切除術后于外院接受6周期異環磷酰胺聯合阿霉素化療。于我院病理會診明確診斷為FS-DFSP后,臨床建議患者行伊馬替尼靶向治療,患者未接受,最終選擇使用安羅替尼聯合艾立布林化療方案。治療期間復查,腹腔包塊切除術后15個月后全腹CT檢查顯示:左側腹盆腔多發結節腫塊影,左下腹腔較大者約8.4 cm×5.3 cm×3.3 cm(圖1l),增強掃描明顯強化;左側腹壁平臍層面稍上處皮下見扁平軟組織密度結節影,大小2.5 cm×0.7 cm,增強掃描可見中度強化;左髖外后方環狀強化軟組織腫塊,大小約3.5 cm×2.4 cm,考慮腫瘤(復發/轉移)。自左側會陰部包塊切除術至今,患者帶病生存149個月。
1.5 文獻復習
通過以“纖維肉瘤型隆突性皮膚纖維肉瘤”“轉移”“fibrosarcomatous dermatofibrosarcoma protuberans”和“metastasis”為檢索詞,檢索PubMed、Embase、Web of Science、CNKI、維普及萬方數據庫,至2024年8月,檢索并篩選出報道單一腹腔轉移性FS-DFSP的文獻6篇,病例共6例[3-8]。見補充材料1。
6例患者的發病年齡為20~60歲(中位年齡為49歲),以男性居多(5/6)。在具有臨床資料的病例中,原發腫瘤發生于軀干4例(包括腹部2例、背部和肩部各1例)、大腿1例,1例不詳。臨床主要表現為生長緩慢的體表腫塊。原發腫瘤直徑為12.6~14.0 cm;組織學形態包括4例FS-DFSP,2例DFSP(無詳細分型),該2例DFSP未詳細描述形態、免疫組織化學及分子遺傳學特征。
腫瘤轉移至腹腔器官4例(胰腺3例,肝臟1例),單一腹腔轉移僅有2例;轉移腫瘤直徑為2.6 cm~14.2 cm。其中4例可獲得轉移灶形態學資料,腹腔轉移腫瘤均為典型FS-DFSP形態,表現為梭形細胞呈束狀或魚骨樣排列;免疫染色CD34呈弱陽性或彌漫強陽性表達,Ki?67陽性指數為10%~40%;除本病例外,僅有1例進行了分子檢測,檢出COL1A1::PDGFB融合基因。
原發/復發腫瘤的治療方式以手術切除為主(6/6),包括廣泛局部切除(4例)和局部切除(2例),局部切除術后輔以放療1例。轉移腫瘤多行手術切除(3/4),術后聯合靶向治療1例,單獨行化療1例。在預后方面,隨訪時間為36~372個月(中位隨訪時間為60個月)。復發4例,復發次數為1~5次。腫瘤發生轉移的時間為15~366個月(中位轉移時間為48個月)。至文獻發表有3例存活,3例患者因病死亡(死于轉移引起的并發癥)。
2 討論
DFSP是重要的皮膚軟組織惡性腫瘤。當臨床病理特征表現典型時,大多數病理醫師對其診斷并不困難。然而,當該腫瘤的發生部位、形態、免疫組織化學特征不典型時,將給診斷帶來極大挑戰。需要指出的是,DFSP疑難病例的診斷依賴于高年資軟組織腫瘤專科病理醫師的經驗并結合分子檢測手段等進行綜合分析。值得注意的是,在這個精準診療時代,DFSP的精準診斷對于臨床個性化治療具有重要意義。本研究報道了1例發生及轉移部位罕見、形態學及免疫表型不典型的FS-DFSP,并進行了文獻復習,以加強對FS-DFSP疑難病例臨床病理特征的認識。
2.1 DFSP的臨床特征
DFSP好發于30~50歲的青中年人群[9]。其發生部位常見位于軀干和四肢近端,少數發生于頭頸[10]。結合臨床,本病例腫瘤原發于會陰,而后腫瘤復發,廣泛浸潤累及髖部,發生遠處轉移形成腹腔包塊。根據文獻[11]報道,極少數DFSP(約0.5%)可發生于生殖系統,包括外陰、陰囊、陰莖等部位。因此,需警惕發生于罕見部位的DFSP給診斷帶來的困難。還需注意的是,詳盡了解患者的臨床信息可能有助于診斷。需要指出的是,近年來發現于子宮的罕見肉瘤亞型,組織學及基因特征與DFSP一致,被稱為COL1A1::PDGFB融合的子宮肉瘤,有學者認為其本質上與DFSP為近親腫瘤[12]。
2.2 DFSP的組織學形態及免疫組織化學特征
經典型DFSP的組織學形態表現為溫和一致的梭形腫瘤細胞呈席紋狀、旋渦樣排列。免疫組織化學染色中腫瘤細胞彌漫強陽性表達CD34。因此,對于侵襲性及低度惡性的梭形細胞腫瘤,當CD34表達同時其他標記陰性是DFSP重要的診斷線索。需要注意的是,經典型DFSP轉化為纖維肉瘤亞型后,形態通常表現為異型性明顯增加,核分裂象活躍。在纖維肉瘤成分中CD34表達可減弱甚至缺失,可能增加診斷難度[13]。值得指出的是,本例轉移灶部位為腹腔,極其罕見,且組織形態表現為明顯的黏液樣變,同時CD34表達陰性,未表現典型的纖維肉瘤特征。僅憑上述特征難以考慮到該腫瘤為FS-DFSP,易誤診為其他高級別肉瘤,因此診斷難度極大。
2.3 DFSP的分子遺傳學特征
細胞遺傳學上,超過90%的DFSP具有特征性COL1A1::PDGFB融合基因。因此通過FISH技術,使用PDGFB斷裂探針和COL1A1::PDGFB融合探針聯合檢測融合基因相關改變,對于DFSP的輔助診斷以及指導臨床靶向治療具有重要意義。在本病例中,腫瘤發生及轉移部位罕見、組織形態困難以及免疫表型不典型,導致難以選擇FS-DFSP相關的FISH檢測項目輔助診斷,由此更強調了疑難病例經驗積累的重要性。還需特別指出的是,有極少部分DFSP(約4%)可能存在COL1A1::PDGFB隱匿性融合、PDGFD重排或其他罕見的分子改變,在FISH雙探針聯合檢測中呈陰性,稱為分子未確定型DFSP[13]。對于此類病例,需補充其他分子檢測手段如二代測序等進一步分析。
纖維肉瘤轉化是DFSP預后最重要的預測因素,其中的分子機制仍不明確。近年來研究發現CDKN2A/2B缺失[14]、12q15基因擴增[15]、P53突變[16, 17]及PDGFRβ/AKT/mTOR通路改變[18]等因素可能在不同程度上與纖維肉瘤轉化相關。本例FS-DFSP中未檢出CDKN2A/2B基因缺失,但可能存在其他遺傳學改變,提示我們在日常病理診斷工作中可通過檢測相關遺傳學異常來關注FS-DFSP患者的預后情況。
2.4 鑒別診斷
本病例首診分類未清楚,其不典型的臨床病理和免疫組織化學特征極易誤診為其他軟組織腫瘤,給準確診斷及患者個性化治療帶來挑戰。該病例需要與好發于腹腔、腹膜后含黏液樣變的高級別腫瘤進行鑒別,主要包括以下幾種類型。
2.4.1 去分化脂肪肉瘤
去分化脂肪肉瘤(dedifferentiated liposarcoma,DDLPS)好發于腹腔和腹膜后。需注意的是,缺乏高分化成分的DDLPS可導致診斷困難。DDLPS具有標志性12q13-15擴增,可運用免疫組織化學表達MDM2、CDK4等標記鑒別,FISH檢測出MDM2、CDK4、FRS2等基因擴增而COL1A1::PDGFB融合基因陰性可輔助鑒別。值得指出的是,MDM2基因擴增可偶見于其他類型軟組織腫瘤(如橫紋肌肉瘤等),而在DFSP中罕見,此特征可能帶來新的診斷陷阱。本課題組[15]曾報道1例極罕見的FS-DFSP伴有MDM2基因擴增。對該類疑難病例,需緊密聯系病史,以形態學為基礎,結合免疫表型及基因檢測結果綜合分析,辯證思考。
2.4.2 黏液樣脂肪肉瘤
黏液樣脂肪肉瘤(myxoid liposarcoma,MLPS)好發于肢體深部軟組織,偶可見轉移至腹腔。腫瘤具有顯著的黏液背景,間質內可見特征性“雞爪樣”毛細血管,腫瘤細胞呈短梭形或卵圓形。腫瘤細胞不同程度表達S100,不表達CD34。FISH檢測DDIT3基因重排可協助診斷。
2.4.3 胃腸道間質瘤
胃腸道間質瘤(gastrointestinal stromal tumor,GIST)是最重要的腹腔肉瘤之一,有時伴有黏液樣改變,腫瘤細胞為形態一致的圓形細胞,呈片狀分布。部分腫瘤免疫組化可見CD34表達陽性,DOG1和CD117呈陰性。85%以上的病例可通過檢測KIT及PDGFRA基因突變協助鑒別。
2.4.4 孤立性纖維性腫瘤
孤立性纖維性腫瘤(solitary fibrous tumor,SFT)是重要的腹腔肉瘤,常有血管外皮瘤樣改變,部分可有黏液樣變性,與本例部分區域形態相似。免疫組織化學可輔助診斷,SFT可表達CD34,同時表達STAT6。SFT中高頻存在NAB2::STAT6融合基因,困難病例可通過RT-PCR或二代測序檢測該融合。
2.4.5 惡性外周神經鞘膜瘤
惡性外周神經鞘膜瘤(malignant peripheral nerve sheath tumor,MPNST)可發生于任何部位,包括腹腔。間質可呈黏液樣至膠原樣,梭形腫瘤細胞常呈條束狀、魚骨樣排列。H3K27me3表達缺失是重要的診斷依據。NF1、CDKN2A/2B、PRC2核心組件(EED或SUZ12)突變是MPNST重要的驅動基因變異,在疑難病例中可結合二代測序綜合分析。
2.4.6 NTRK重排梭形細胞腫瘤
該腫瘤是近年來新分類的一組罕見軟組織腫瘤,少數可發生于腹盆腔。腫瘤形態多樣,部分由具有異型性的形態一致的梭形細胞構成,呈條束狀或魚骨狀排列,免疫組織化學可表達CD34,且pan-TRK表達陽性。可通過FISH檢測NTRK1/2/3基因重排幫助鑒別。
2.5 DFSP的治療及預后
DFSP的主要治療方式是手術切除,對傳統化療藥物反應性差。基于DFSP的腫瘤發生機制,靶向PDGFR位點的酪氨酸激酶抑制劑伊馬替尼成為治療研究的熱點。伊馬替尼在GIST中顯示出了良好的治療效果,目前也被批準應用于難治性/復發性/轉移性DFSP的治療,已觀察到具有潛力的療效。雖然本例腹腔轉移性FS-DFSP患者未接受伊馬替尼靶向治療,但其已明確為COL1A1::PDGFB融合陽性病例,存在50%的可能對伊馬替尼產生應答。
綜上,本文報道了1例腹腔轉移性FS-DFSP,其具有罕見的發生/轉移部位、不典型的組織學形態和免疫表型,為診斷帶來多重陷阱,使之不易被考慮為FS-DFSP,導致難以選擇合適的分子檢測項目對其進行精準診斷。因此,應加深病理及臨床醫師對該類腫瘤的認識,在臨床病理工作中,以組織形態作為診斷的基石,詳細了解臨床病史,合理選擇免疫組織化學及分子檢測,有益于對DFSP患者進行準確的診斷及制定個性化的治療方案。
重要聲明
利益沖突聲明:所有作者聲明無利益沖突。
作者貢獻聲明:崔賢鎮負責查閱相關文獻、數據收集與分析,撰寫文章;陸楊負責文獻及數據的核對,以及文章修改;何鑫負責文章修改;張紅英負責病例診斷、指導文章寫作和修改。
倫理聲明:本研究已通過四川大學華西醫院生物醫學倫理審查委員會的倫理審核批準,批文編號:2023年審(1374)號。
隆突性皮膚纖維肉瘤(dermatofibrosarcoma protuberans,DFSP)是一種發生于淺表的局部侵襲性纖維母細胞腫瘤,具有標志性COL1A1::PDGFB融合基因或其他相關融合[1]。經典型DFSP為低度惡性腫瘤,有10%~16%的經典型DFSP可通過高級別轉化形成纖維肉瘤型DFSP(fibrosarcomatous DFSP,FS-DFSP),生物學行為表現為侵襲性增強,具備遠處轉移潛能[2]。該腫瘤最常見的轉移部位為肺,還可見轉移至腦、骨等,單一腹腔轉移性FS-DFSP非常罕見。本研究報道了四川大學華西醫院(簡稱“我院”)于2023年診斷的1例腹腔轉移性FS-DFSP,該病例在診斷上極具挑戰性,現將其臨床病理資料及診治經過進行回顧,并結合國內外文獻報道進行分析,以期提高對疑難病例的認識、診斷及鑒別診斷水平。
1 臨床資料
1.1 一般資料
患者,女,51歲,因“反復左下腹痛6個月”于當地醫院就診。10年前因左側會陰部包塊行手術切除,外院診斷為“皮下纖維性腫瘤”,未能進一步分型。
1.2 外院診斷及處理
查體:左髖部捫及包塊,觸診大小約14 cm×10 cm×8 cm,皮膚表面張力高,見靜脈充盈明顯,皮溫較對側高,包塊表面光滑、質韌、有壓痛、與周圍組織邊界不清、活動度差。輔助檢查:CA125 59.3 U/mL(正常參考值<35.0 U/mL),CA72-4 8.0 U/mL(正常參考值<6.9 U/mL)。影像學檢查:① 腹部增強MRI檢查示腹腔左側一類圓形巨大腫塊,大小約14.2 cm×11.7 cm,T1WI呈稍低信號及部分為高信號,T2WI呈高信號伴散在少許低信號,增強掃描實性部分呈持續性不均勻強化,病灶與脾門區結構分界不清,胰腺受壓后移,胃及左腎受壓變形、移位,考慮惡性腫瘤可能性大。② 骨盆平掃MRI檢查示左側髖部肌間隙內不規則腫塊影,大小約14.0 cm×11.0 cm×5.8 cm,T1WI以等信號為主,其內散在點狀高信號影,T2信號混雜,以等、稍高信號為主,邊界欠清晰,鄰近臀大肌稍腫脹,臀大肌、臀小肌受壓推擠,考慮惡性腫瘤可能性大。行超聲引導下腹腔包塊穿刺活檢,病理診斷考慮脂肪源性肉瘤。臨床首先行腹腔包塊切除,術中見:腹腔左側一巨大腫塊,大小約14.2 cm×12.0 cm×10.0 cm,包膜較完整,腫瘤周圍與胃、結腸、脾及胰體尾粘連。大體見灰褐色包塊1個,大小13.0 cm×12.0 cm×9.5 cm,包塊表面與胰腺粘連,似有包膜,切面灰白灰褐,質嫩,局部似有出血,呈囊性變,囊內含血凝塊樣物。當地醫院病理診斷:“腹腔梭形細胞惡性腫瘤,類型不能除外上皮樣血管內皮瘤或去分化脂肪肉瘤”。患者為進一步明確腫瘤類型,于2023年8月至我院進行病理會診。
腹腔手術1個月后患者行髖部包塊切除,當地醫院病理診斷:“髖部梭形細胞惡性腫瘤”。患者將髖部包塊病理切片送至我院進行病理會診。
1.3 我院病理會診情況
腹腔腫物鏡下可見腫瘤邊界清楚(圖1a),間質疏松,繼發黏液樣變(圖1b),其中含較多薄壁小血管,局灶可見血管外皮瘤樣構象(圖1c)。腫瘤主要由中度以上異型性的短梭形或卵圓形細胞組成,呈不規則排列(圖1d)。細胞核染色質呈粗顆粒狀,核仁不明顯,胞質呈嗜酸性。核分裂象易見(約10個/10 HPF),會診切片未見出血壞死。免疫組織化學結果顯示,腫瘤細胞CD34(圖1e)、結蛋白(Desmin)、S100、CD117和DOG1均為陰性,Ki?67陽性指數約為15%。熒光原位雜交(fluorescence in situ hybridization,FISH)檢測檢出PDGFB基因不平衡重排(圖1f)及COL1A1::PDGFB基因融合(圖1g),未檢出MDM2基因擴增、未檢出DDIT3基因重排。病理診斷為腹腔軟組織惡性腫瘤(肉瘤),結合免疫組織化學及基因檢測結果,亞型診斷為FS-DFSP;FNCLCC(法國國家癌癥中心聯盟)分級為2級。本病例首先需排除腹腔轉移性腫瘤,應積極查找原發病灶。
圖1
示該例患者的腹腔腫瘤和髖部腫瘤的組織學、免疫表型和分子遺傳學特征,以及明確FS-DFSP診斷后治療期間的CT復查結果
a~g為腹腔腫瘤結果;h~l為髖部腫瘤結果;a:腹腔腫瘤呈結節狀,邊界清楚(HE ×200);b:腫瘤間質疏松,繼發黏液樣變,間質內可見豐富的薄壁小血管(HE ×400);c:腫瘤間質部分區域可見血管外皮瘤樣構象(HE ×200);d:腫瘤由短梭形或卵圓形細胞組成,排列不規則,核分裂象活躍(黑箭,HE ×400);e:腹腔腫瘤細胞CD34呈陰性(EnVision法 ×400);f:FISH結果提示PDGFB基因重排(黃箭),PDGFB基因為紅色和綠色信號;g:FISH結果提示存在COL1A1::PDGFB融合基因(黃箭),COL1A1基因為紅色信號,PDGFB基因為綠色信號;h:髖部腫瘤大部分區域形態與腹腔腫瘤相似,由短梭形或卵圓形細胞呈不規則或短束狀結構排列組成(HE ×200);i:髖部腫瘤部分區域可見梭形細胞呈魚骨樣排列(HE ×200);j:髖部腫瘤CD34部分陽性(EnVision法 ×400);k:FISH結果提示存在COL1A1::PDGFB融合基因(黃箭),COL1A1基因為紅色信號,PDGFB基因為綠色信號;l:腹腔包塊切除術后15個月CT圖像示腹腔轉移灶復發,增強掃描明顯強化(黃箭)
髖部包塊鏡下觀察:髖部病變大部分區域形態與腹腔腫瘤相似(圖1h),部分區域可見中-重度異型性的梭形腫瘤細胞,呈束狀或魚骨樣結構排列(圖1i),核分裂象活躍(27個/10 HPF),部分區域可見壞死;腫瘤細胞CD34部分陽性(圖1j),其余標記與腹腔病變一致,信號轉導與轉錄激活因子6(signal transducer and activator of transcription-6,STAT6)和廣譜原肌球蛋白受體激酶(pan-tropomyosin receptor kinase,pan-TRK)均呈陰性,H3K27Me3未缺失;Ki?67陽性指數約為40%。FISH檢測檢出PDGFB基因不平衡重排及COL1A1::PDGFB基因融合(圖1k),未檢出CDKN2A基因缺失。病理診斷為髖部軟組織惡性腫瘤(肉瘤)。結合臨床、形態學、免疫組織化學及基因檢測結果,亞型診斷為FS-DFSP,支持髖部包塊與腹腔包塊為同源性腫瘤。
經臨床及病理軟組織專科多學科會診,聯系臨床,發現髖部腫瘤切除范圍大,與會陰部手術范圍相連。結合病理特征分析,考慮髖部腫瘤系會陰部腫瘤廣泛浸潤累及所致,腹腔腫瘤為轉移性FS-DFSP。
1.4 治療及隨訪
患者髖部包塊切除術后于外院接受6周期異環磷酰胺聯合阿霉素化療。于我院病理會診明確診斷為FS-DFSP后,臨床建議患者行伊馬替尼靶向治療,患者未接受,最終選擇使用安羅替尼聯合艾立布林化療方案。治療期間復查,腹腔包塊切除術后15個月后全腹CT檢查顯示:左側腹盆腔多發結節腫塊影,左下腹腔較大者約8.4 cm×5.3 cm×3.3 cm(圖1l),增強掃描明顯強化;左側腹壁平臍層面稍上處皮下見扁平軟組織密度結節影,大小2.5 cm×0.7 cm,增強掃描可見中度強化;左髖外后方環狀強化軟組織腫塊,大小約3.5 cm×2.4 cm,考慮腫瘤(復發/轉移)。自左側會陰部包塊切除術至今,患者帶病生存149個月。
1.5 文獻復習
通過以“纖維肉瘤型隆突性皮膚纖維肉瘤”“轉移”“fibrosarcomatous dermatofibrosarcoma protuberans”和“metastasis”為檢索詞,檢索PubMed、Embase、Web of Science、CNKI、維普及萬方數據庫,至2024年8月,檢索并篩選出報道單一腹腔轉移性FS-DFSP的文獻6篇,病例共6例[3-8]。見補充材料1。
6例患者的發病年齡為20~60歲(中位年齡為49歲),以男性居多(5/6)。在具有臨床資料的病例中,原發腫瘤發生于軀干4例(包括腹部2例、背部和肩部各1例)、大腿1例,1例不詳。臨床主要表現為生長緩慢的體表腫塊。原發腫瘤直徑為12.6~14.0 cm;組織學形態包括4例FS-DFSP,2例DFSP(無詳細分型),該2例DFSP未詳細描述形態、免疫組織化學及分子遺傳學特征。
腫瘤轉移至腹腔器官4例(胰腺3例,肝臟1例),單一腹腔轉移僅有2例;轉移腫瘤直徑為2.6 cm~14.2 cm。其中4例可獲得轉移灶形態學資料,腹腔轉移腫瘤均為典型FS-DFSP形態,表現為梭形細胞呈束狀或魚骨樣排列;免疫染色CD34呈弱陽性或彌漫強陽性表達,Ki?67陽性指數為10%~40%;除本病例外,僅有1例進行了分子檢測,檢出COL1A1::PDGFB融合基因。
原發/復發腫瘤的治療方式以手術切除為主(6/6),包括廣泛局部切除(4例)和局部切除(2例),局部切除術后輔以放療1例。轉移腫瘤多行手術切除(3/4),術后聯合靶向治療1例,單獨行化療1例。在預后方面,隨訪時間為36~372個月(中位隨訪時間為60個月)。復發4例,復發次數為1~5次。腫瘤發生轉移的時間為15~366個月(中位轉移時間為48個月)。至文獻發表有3例存活,3例患者因病死亡(死于轉移引起的并發癥)。
2 討論
DFSP是重要的皮膚軟組織惡性腫瘤。當臨床病理特征表現典型時,大多數病理醫師對其診斷并不困難。然而,當該腫瘤的發生部位、形態、免疫組織化學特征不典型時,將給診斷帶來極大挑戰。需要指出的是,DFSP疑難病例的診斷依賴于高年資軟組織腫瘤專科病理醫師的經驗并結合分子檢測手段等進行綜合分析。值得注意的是,在這個精準診療時代,DFSP的精準診斷對于臨床個性化治療具有重要意義。本研究報道了1例發生及轉移部位罕見、形態學及免疫表型不典型的FS-DFSP,并進行了文獻復習,以加強對FS-DFSP疑難病例臨床病理特征的認識。
2.1 DFSP的臨床特征
DFSP好發于30~50歲的青中年人群[9]。其發生部位常見位于軀干和四肢近端,少數發生于頭頸[10]。結合臨床,本病例腫瘤原發于會陰,而后腫瘤復發,廣泛浸潤累及髖部,發生遠處轉移形成腹腔包塊。根據文獻[11]報道,極少數DFSP(約0.5%)可發生于生殖系統,包括外陰、陰囊、陰莖等部位。因此,需警惕發生于罕見部位的DFSP給診斷帶來的困難。還需注意的是,詳盡了解患者的臨床信息可能有助于診斷。需要指出的是,近年來發現于子宮的罕見肉瘤亞型,組織學及基因特征與DFSP一致,被稱為COL1A1::PDGFB融合的子宮肉瘤,有學者認為其本質上與DFSP為近親腫瘤[12]。
2.2 DFSP的組織學形態及免疫組織化學特征
經典型DFSP的組織學形態表現為溫和一致的梭形腫瘤細胞呈席紋狀、旋渦樣排列。免疫組織化學染色中腫瘤細胞彌漫強陽性表達CD34。因此,對于侵襲性及低度惡性的梭形細胞腫瘤,當CD34表達同時其他標記陰性是DFSP重要的診斷線索。需要注意的是,經典型DFSP轉化為纖維肉瘤亞型后,形態通常表現為異型性明顯增加,核分裂象活躍。在纖維肉瘤成分中CD34表達可減弱甚至缺失,可能增加診斷難度[13]。值得指出的是,本例轉移灶部位為腹腔,極其罕見,且組織形態表現為明顯的黏液樣變,同時CD34表達陰性,未表現典型的纖維肉瘤特征。僅憑上述特征難以考慮到該腫瘤為FS-DFSP,易誤診為其他高級別肉瘤,因此診斷難度極大。
2.3 DFSP的分子遺傳學特征
細胞遺傳學上,超過90%的DFSP具有特征性COL1A1::PDGFB融合基因。因此通過FISH技術,使用PDGFB斷裂探針和COL1A1::PDGFB融合探針聯合檢測融合基因相關改變,對于DFSP的輔助診斷以及指導臨床靶向治療具有重要意義。在本病例中,腫瘤發生及轉移部位罕見、組織形態困難以及免疫表型不典型,導致難以選擇FS-DFSP相關的FISH檢測項目輔助診斷,由此更強調了疑難病例經驗積累的重要性。還需特別指出的是,有極少部分DFSP(約4%)可能存在COL1A1::PDGFB隱匿性融合、PDGFD重排或其他罕見的分子改變,在FISH雙探針聯合檢測中呈陰性,稱為分子未確定型DFSP[13]。對于此類病例,需補充其他分子檢測手段如二代測序等進一步分析。
纖維肉瘤轉化是DFSP預后最重要的預測因素,其中的分子機制仍不明確。近年來研究發現CDKN2A/2B缺失[14]、12q15基因擴增[15]、P53突變[16, 17]及PDGFRβ/AKT/mTOR通路改變[18]等因素可能在不同程度上與纖維肉瘤轉化相關。本例FS-DFSP中未檢出CDKN2A/2B基因缺失,但可能存在其他遺傳學改變,提示我們在日常病理診斷工作中可通過檢測相關遺傳學異常來關注FS-DFSP患者的預后情況。
2.4 鑒別診斷
本病例首診分類未清楚,其不典型的臨床病理和免疫組織化學特征極易誤診為其他軟組織腫瘤,給準確診斷及患者個性化治療帶來挑戰。該病例需要與好發于腹腔、腹膜后含黏液樣變的高級別腫瘤進行鑒別,主要包括以下幾種類型。
2.4.1 去分化脂肪肉瘤
去分化脂肪肉瘤(dedifferentiated liposarcoma,DDLPS)好發于腹腔和腹膜后。需注意的是,缺乏高分化成分的DDLPS可導致診斷困難。DDLPS具有標志性12q13-15擴增,可運用免疫組織化學表達MDM2、CDK4等標記鑒別,FISH檢測出MDM2、CDK4、FRS2等基因擴增而COL1A1::PDGFB融合基因陰性可輔助鑒別。值得指出的是,MDM2基因擴增可偶見于其他類型軟組織腫瘤(如橫紋肌肉瘤等),而在DFSP中罕見,此特征可能帶來新的診斷陷阱。本課題組[15]曾報道1例極罕見的FS-DFSP伴有MDM2基因擴增。對該類疑難病例,需緊密聯系病史,以形態學為基礎,結合免疫表型及基因檢測結果綜合分析,辯證思考。
2.4.2 黏液樣脂肪肉瘤
黏液樣脂肪肉瘤(myxoid liposarcoma,MLPS)好發于肢體深部軟組織,偶可見轉移至腹腔。腫瘤具有顯著的黏液背景,間質內可見特征性“雞爪樣”毛細血管,腫瘤細胞呈短梭形或卵圓形。腫瘤細胞不同程度表達S100,不表達CD34。FISH檢測DDIT3基因重排可協助診斷。
2.4.3 胃腸道間質瘤
胃腸道間質瘤(gastrointestinal stromal tumor,GIST)是最重要的腹腔肉瘤之一,有時伴有黏液樣改變,腫瘤細胞為形態一致的圓形細胞,呈片狀分布。部分腫瘤免疫組化可見CD34表達陽性,DOG1和CD117呈陰性。85%以上的病例可通過檢測KIT及PDGFRA基因突變協助鑒別。
2.4.4 孤立性纖維性腫瘤
孤立性纖維性腫瘤(solitary fibrous tumor,SFT)是重要的腹腔肉瘤,常有血管外皮瘤樣改變,部分可有黏液樣變性,與本例部分區域形態相似。免疫組織化學可輔助診斷,SFT可表達CD34,同時表達STAT6。SFT中高頻存在NAB2::STAT6融合基因,困難病例可通過RT-PCR或二代測序檢測該融合。
2.4.5 惡性外周神經鞘膜瘤
惡性外周神經鞘膜瘤(malignant peripheral nerve sheath tumor,MPNST)可發生于任何部位,包括腹腔。間質可呈黏液樣至膠原樣,梭形腫瘤細胞常呈條束狀、魚骨樣排列。H3K27me3表達缺失是重要的診斷依據。NF1、CDKN2A/2B、PRC2核心組件(EED或SUZ12)突變是MPNST重要的驅動基因變異,在疑難病例中可結合二代測序綜合分析。
2.4.6 NTRK重排梭形細胞腫瘤
該腫瘤是近年來新分類的一組罕見軟組織腫瘤,少數可發生于腹盆腔。腫瘤形態多樣,部分由具有異型性的形態一致的梭形細胞構成,呈條束狀或魚骨狀排列,免疫組織化學可表達CD34,且pan-TRK表達陽性。可通過FISH檢測NTRK1/2/3基因重排幫助鑒別。
2.5 DFSP的治療及預后
DFSP的主要治療方式是手術切除,對傳統化療藥物反應性差。基于DFSP的腫瘤發生機制,靶向PDGFR位點的酪氨酸激酶抑制劑伊馬替尼成為治療研究的熱點。伊馬替尼在GIST中顯示出了良好的治療效果,目前也被批準應用于難治性/復發性/轉移性DFSP的治療,已觀察到具有潛力的療效。雖然本例腹腔轉移性FS-DFSP患者未接受伊馬替尼靶向治療,但其已明確為COL1A1::PDGFB融合陽性病例,存在50%的可能對伊馬替尼產生應答。
綜上,本文報道了1例腹腔轉移性FS-DFSP,其具有罕見的發生/轉移部位、不典型的組織學形態和免疫表型,為診斷帶來多重陷阱,使之不易被考慮為FS-DFSP,導致難以選擇合適的分子檢測項目對其進行精準診斷。因此,應加深病理及臨床醫師對該類腫瘤的認識,在臨床病理工作中,以組織形態作為診斷的基石,詳細了解臨床病史,合理選擇免疫組織化學及分子檢測,有益于對DFSP患者進行準確的診斷及制定個性化的治療方案。
重要聲明
利益沖突聲明:所有作者聲明無利益沖突。
作者貢獻聲明:崔賢鎮負責查閱相關文獻、數據收集與分析,撰寫文章;陸楊負責文獻及數據的核對,以及文章修改;何鑫負責文章修改;張紅英負責病例診斷、指導文章寫作和修改。
倫理聲明:本研究已通過四川大學華西醫院生物醫學倫理審查委員會的倫理審核批準,批文編號:2023年審(1374)號。