

引用本文: 徐瑤, 黃靚, 周有峰, 胡春輝. PIGO基因變異相關發育性癲癇性腦病不伴堿性磷酸酶增高一例并文獻復習. 癲癇雜志, 2024, 10(6): 528-532. doi: 10.7507/2096-0247.202408002 復制
磷脂酰肌醇聚糖錨定生物合成O類蛋白(phosphatidylinositol glycan anchor biosynthesis class O protein,PIGO)基因編碼糖基磷脂酰肌醇-乙醇胺磷酸轉移酶3,參與糖基磷脂酰肌醇(glycosylphosphatidylinositol,GPI)錨定合成途徑的重要步驟,用于將蛋白質附著于細胞表面,維持正常神經元和胚胎發育[1]。目前報道,PIGO基因變異相關疾病存在兩種表型,一種為伴有精神發育遲滯綜合征(hyperphosphatasia with mental retardation syndrome,HPMRS/Mabry syndrome)的高磷酸酯酶癥,是一種常染色體隱性遺傳的智力障礙,其特征為面容畸形、發育遲緩和血清堿性磷酸酶持續升高(高磷酸鹽血癥)[2,3]。PIGO基因變異另一種表型為發育性癲癇性腦病,可伴胼胝體發育異常,伴或不伴輕度堿性磷酸酶增高[4]。本研究通過總結福建省兒童醫院神經內科2023年8月收治的1例患兒的臨床特點,并文獻復習,提高臨床認識。本研究經福建省兒童醫院醫學倫理委員會審核批準(2023ETKLR11006)及患兒監護人知情同意。
病例資料 患兒 男,6月齡20天,因“間斷抽搐5 h余”入院。患兒于入院前5 h于家中洗澡后出現抽搐發作,表現為意識喪失,雙眼正前方凝視,右側眼瞼頻繁眨動,口唇青紫,持續約1 h緩解,緩解后發作間歇期意識不清,間隔半小時后再發抽搐,發作形式基本同前,偶伴有不自主尖叫,抽搐時體溫38℃,就診當地醫院予苯巴比妥、地西泮止驚,期間血氧飽和度下降,最低降至78%,予氣管插管呼吸機輔助機械通氣,遂轉入我院,轉診途中仍有間斷抽搐,表現同前,間斷持續約5 h余至我院。患兒系G1P1,足月剖宮產,出生體重3.6 kg,出生史無異常,生后母乳喂養,新生兒期因“病理性黃疸”住院治療,入院前9天因“支氣管肺炎”在當地醫院住院治療。患兒生長發育較同齡兒明顯落后,目前豎頭不穩,不會翻身,不能追聲追物。4月齡開始一直行康復訓練,發育未見進步。父母健康,非近親結婚,無類似疾病家族遺傳史。入院查體:氣管插管呼吸機輔助通氣鎮靜狀態,營養不良,雙肺呼吸音粗,未聞及干濕啰音,心臟腹部查體無異常。四肢自主活動少,四肢肌張力低,肌力查體不配合。跟腱、膝腱反射可引出。腦膜刺激征陰性,病理征陰性。入院輔助檢查:血常規、血生化、血清堿性磷酸酶、腦脊液常規、生化、培養、甲功、凝血、血尿代謝篩查等均大致正常。視頻腦電圖提示醒睡各期雙側頂枕顳區棘波、棘慢波、尖形慢波、不規則慢波發放。頭顱核磁提示雙側側腦室增寬,胼胝體發育不良(圖1)。入院后予左乙拉西坦口服抗癲癇發作等對癥支持治療,逐漸撤離呼吸機,住院治療10天好轉后出院。出院間隔1個月后再發抽搐,發作形式基本同前,頻率較前增多,發作頻繁時每天數次,容易出現癲癇持續狀態,多數持續10 min以上,應用咪達唑侖口頰粘膜溶液止驚后發作可終止。后加用丙戊酸口服抗癲癇發作治療,抽搐頻率較前下降,多在感染等情況下誘發發作。后行基因全外顯子測序分析,結果顯示PIGO基因復合雜合變異:c.2607_2609del(p.Leu870del)雜合變異,變異來自母親,父親為野生型;c.3070-26A>T雜合變異,變異來自父親,母親為野生型(圖2)。根據ACMG評級、基因多態性、基因保守性、等位基因頻率等評估致病性,使用SIFT、Polyphen HDIV、Polyphen HVAR、LRT、Mutation Taster和Mutation Assessor多種軟件預測,變異均為有害,Splice AI預測c.3070-26A>T變異,預測分值0.58,遠高于常規剪接位點變異的預測閾值,該變異很可能會影響剪接,故綜合評判考慮為可能致病性復合雜合變異,結合文獻報道的該基因的兩種表型,最終該患兒診斷為PIGO基因變異相關發育性癲癇性腦病不伴輕度堿性磷酸酶增高。目前長期口服左乙拉西坦、丙戊酸抗癲癇發作藥物治療,持續隨訪中。
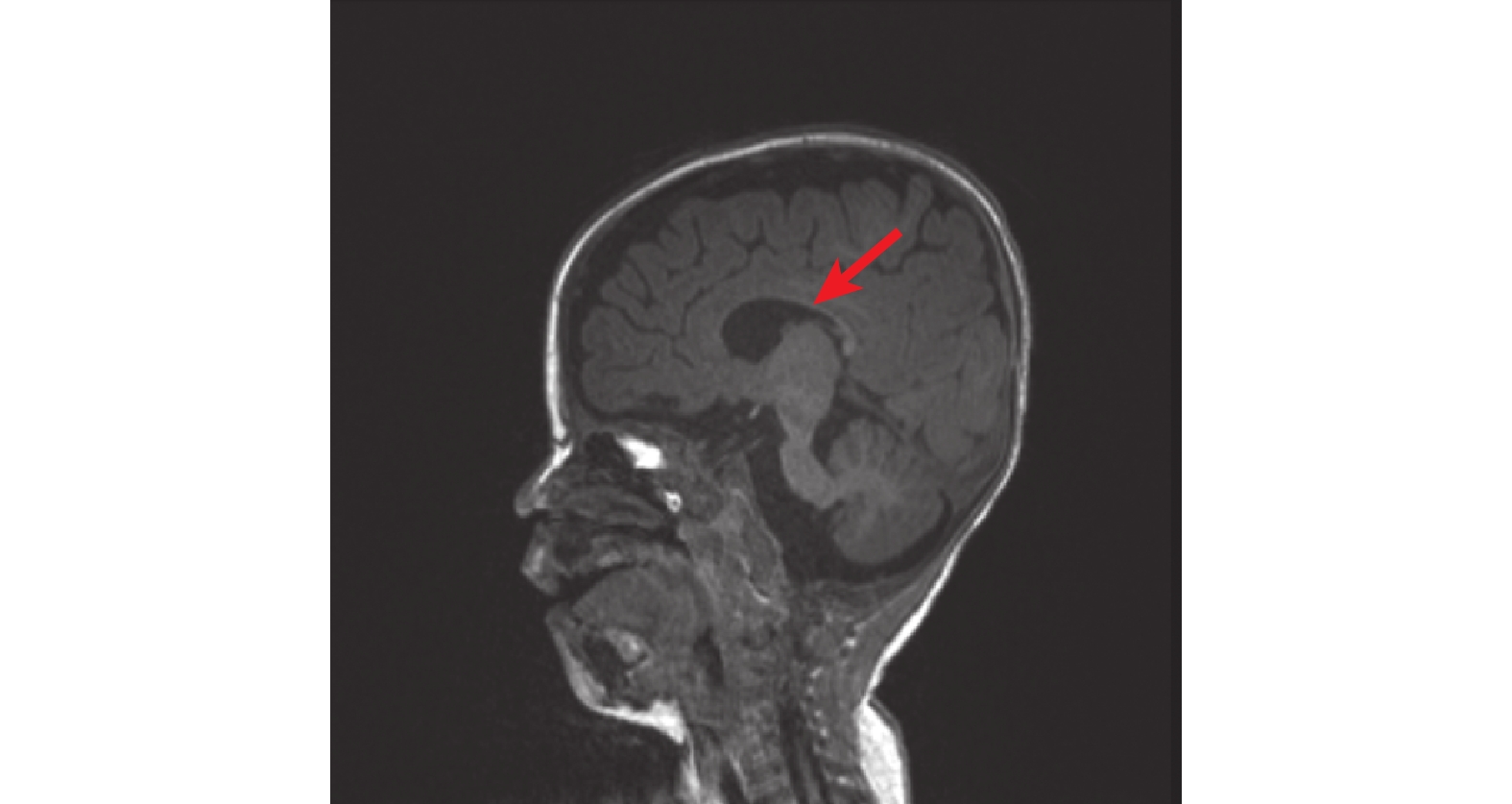
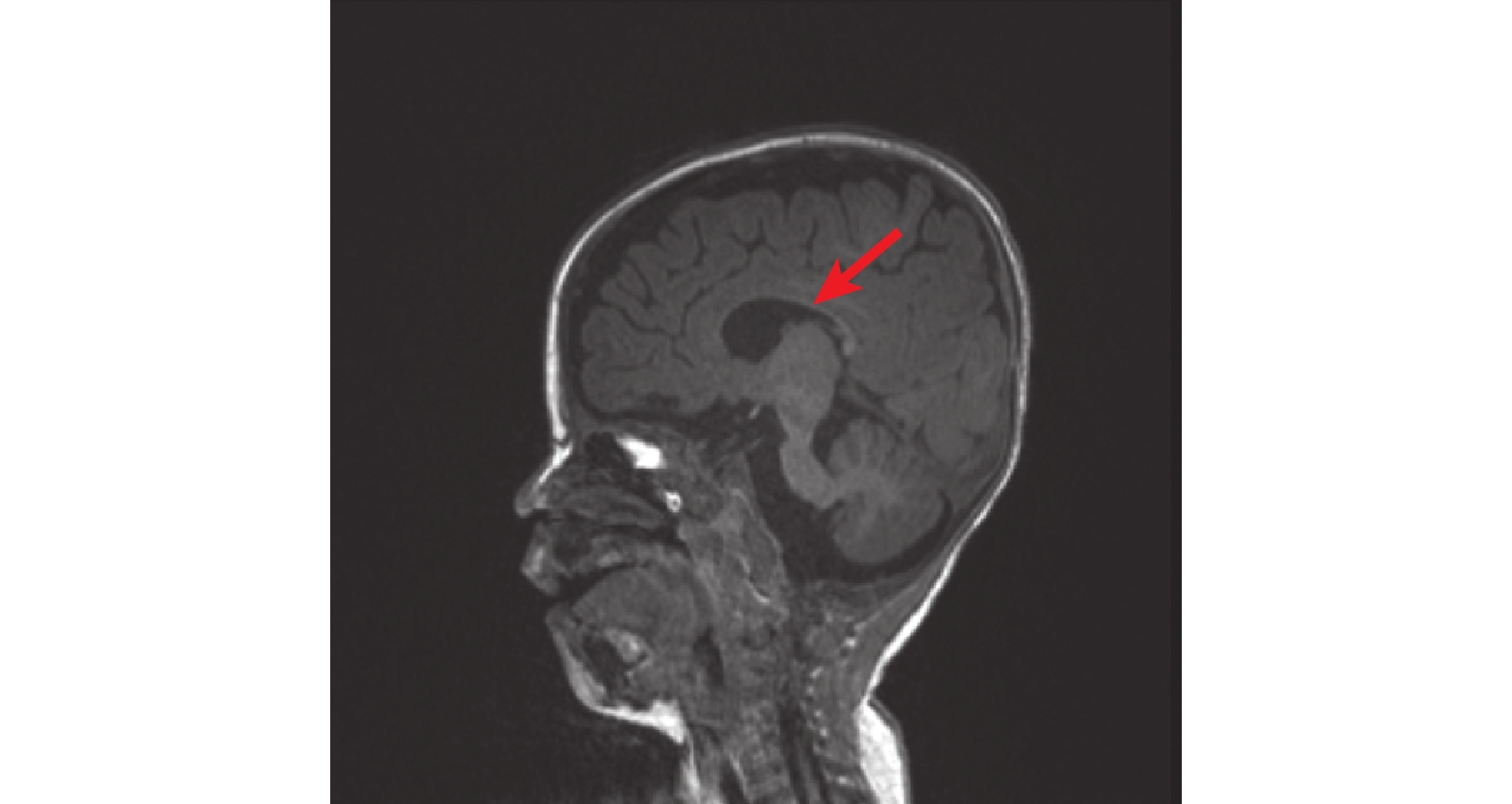 圖1
頭顱核磁共振提示胼胝體發育不良
圖1
頭顱核磁共振提示胼胝體發育不良
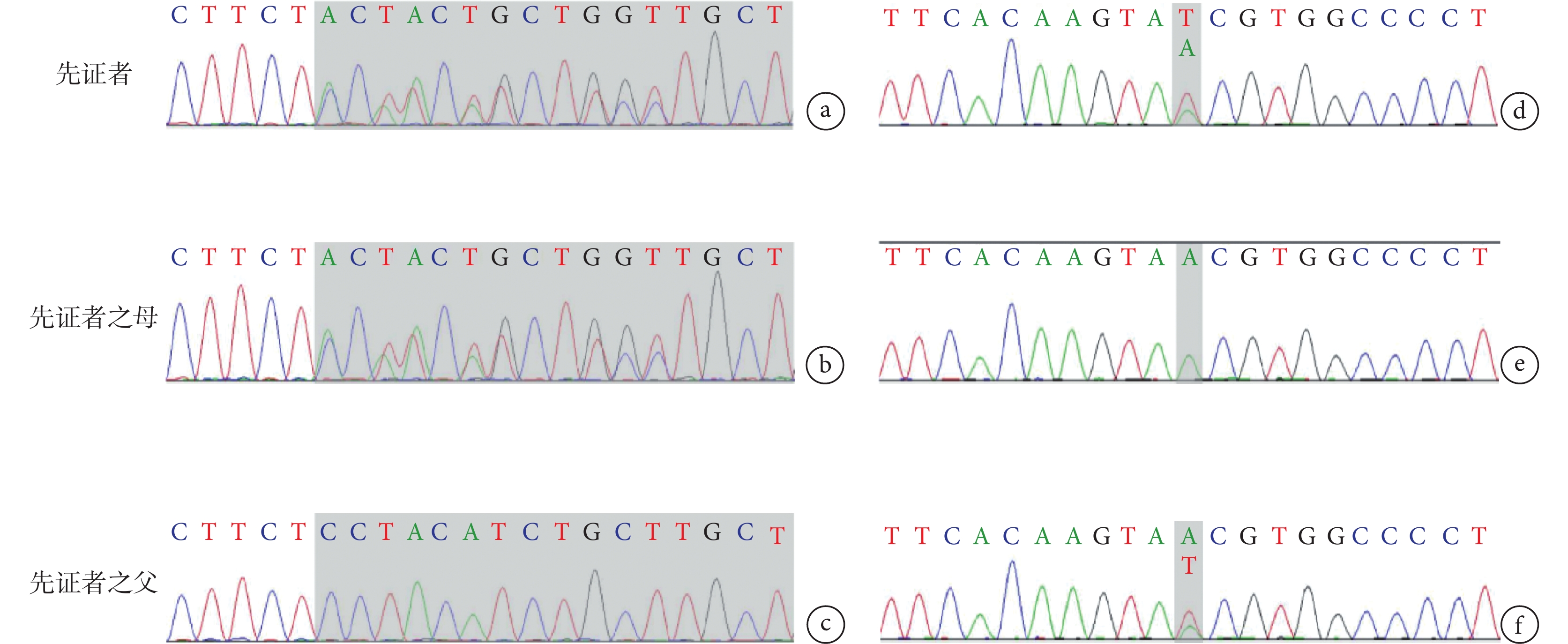
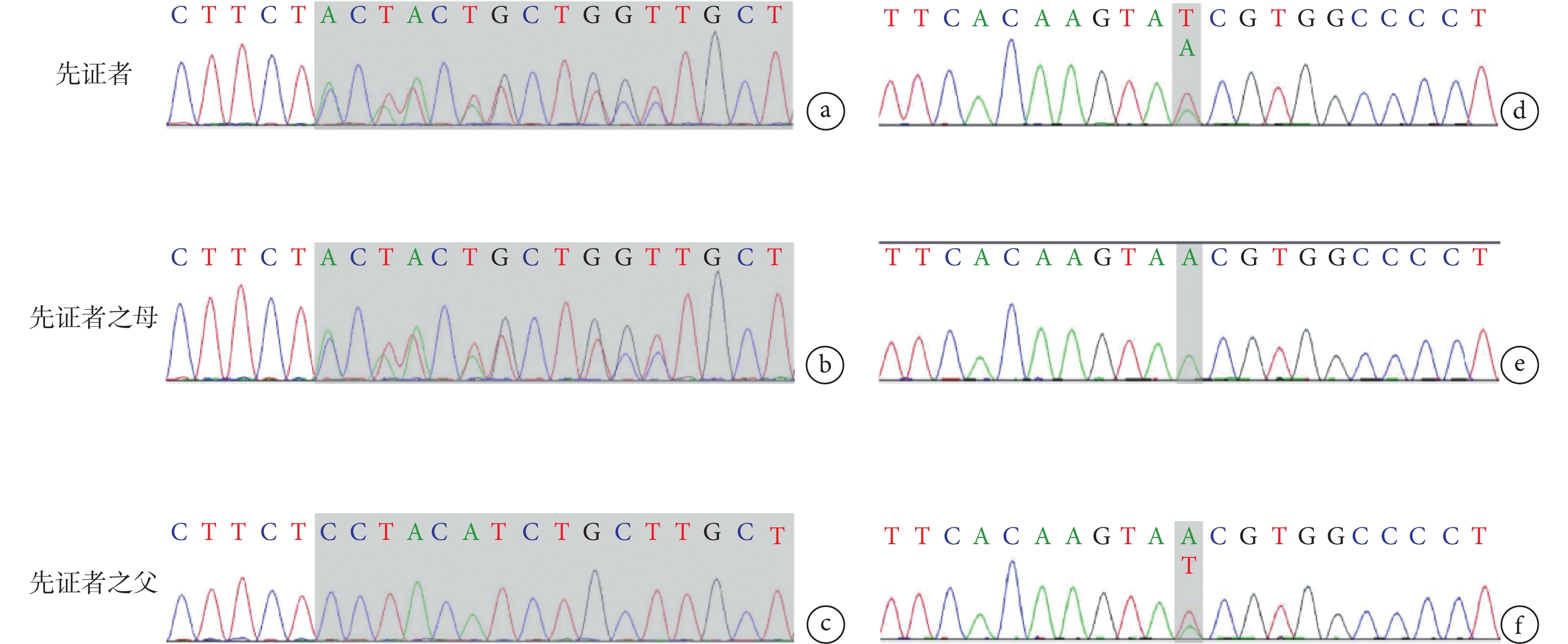 圖2
PIGO基因復合雜合變異
圖2
PIGO基因復合雜合變異
c.2607_2609del(p.Leu870del)雜合變異,變異來自母親,父親為野生型(a,b,c);c.3070-26A>T雜合變異,變異來自父親,母親為野生型(d,e,f)
文獻復習 分別以“PIGO、PIGO mutation、發育性癲癇性腦病、癲癇性腦病”為檢索詞檢索PubMed、中國知網、萬方醫學網等數據庫(建庫至2024年6月),發現檢索符合條件的中文文獻0篇、英文文獻11篇共20例PIGO基因變異相關發育性癲癇性腦病患兒[1-11]。所有報道的患者,年齡9月齡~22歲,均出現全面性發育遲緩或智力障礙(通常為重度),其余臨床特征包括:癲癇發作(13/20)、先天性巨結腸和肛門閉鎖等腸道畸形(12/20)、指畸形(包括短指畸形和指甲發育不全)(10/20)、感音神經性聽力損失(6/20)、舞蹈病(4/20)、先天性泌尿生殖系統畸形(3/20)、先天性心臟病(3/20)和食管閉鎖(2/20)。顱縫早閉和椎體異常各有1例。11/20個體存在面部畸形,常見的有寬瞼裂、高鼻梁、腭裂等。15/20個體存在高磷酸鹽血癥(即血清堿性磷酸酶水平升高)。1例病程中并發脫水、低血糖伴功能性高胰島素血癥、皮質醇減退癥以及橫紋肌溶解癥。神經影像學檢查結果包括腦白質病變(5/20)、腦萎縮伴腦室腫大(5/20)、小腦萎縮(4/20)、胼胝體薄(3/20)、基底神經節或腦干彌散受限(3/20)以及視神經發育不良(1/20)。20例PIGO基因變異位點及對應臨床表型特點總結見表1。
20例患兒中13例出現癲癇發作,癲癇發作起病年齡為4日齡~1歲11月齡,癲癇發作形式包括局灶性發作、強直陣攣發作、全面性強直發作、全面性強直陣攣發作,聯合應用2~4種抗癲癇發作藥物,包括苯巴比妥、拉莫三嗪、托吡酯、左乙拉西坦、丙戊酸、氯巴占、司替戊醇、維生素B6、吡哆醇等,其中2例癲癇發作控制,11例未控制。其中1例使用吡哆醇治療有效,每日口服400 mg吡哆醇(20 mg/kg)一周后,觀察到癲癇發作完全停止,發作間期腦電圖改善,吡哆醇給藥中斷容易引起癲癇再次發作,需長期口服維持。1例添加丙戊酸治療,發作控制。最終6例死亡(6/20,30%),死亡年齡為9月齡~2歲8月齡,其中2例c.1109A>G (p.N370S)、c.2497_2498del (p.A834Cfs)復合雜合變異患兒分別死于17月齡、20月齡,1例c.2869C>T(p.L957P)、c.3069+5G>A復合雜合變異患兒死于22月齡,1例c.765G>A (p.M255I)純合變異患兒死于2歲8月齡,1例c.1352T>G (p.Met451Arg)、c.1392delinsGA (p.Ile464Mfs*42)復合雜合變異患兒死于9月齡,1例c.389C>A (p.Thr130Asn)、c.1288C>T (p.Gln430*) 復合雜合變異患兒死于1歲。6例中2例因驚厥持續狀態死亡。
討論 遺傳性GPI缺乏癥(inherited glycosylphosphatidylinositol deficiencies,IGDs)也稱為IGDs,至少有26個基因參與GPI錨定蛋白的生物合成和運輸,目前已有14個基因突變被確定為常染色體隱性遺傳的IGDs的原因:PIGQ、PIGY、PIGC、PIGL、PIGW、PIGM、PIGV、PIGN、PIGO、PIGG、PIGT、PGAP1、PGAP2和PGAP3。其中,PIGV和PIGO突變是最常見的原因[1,2]。IGDs導致GPI錨定蛋白(GPI-anchored proteins,GPI-Aps)表達降低,影響神經元功能和胚胎發育,參與疾病發生發展。
IGDs的臨床表現多種多樣,其特征包括智力障礙、頑固性癲癇、畸形特征和器官異常,臨床表型嚴重程度也多種多樣。由于IGDs是罕見疾病,表型與基因型的相關性尚未得到很好的定義[2]。本組患兒1例,以全面性發育遲緩、癲癇發作不伴有血清磷酸酶升高為主要表現,與文獻中報道的表型符合。臨床上需要注意的是,該基因變異相關癲癇,容易出現癲癇持續狀態,癲癇持續狀態多為早期死因,囑托家長長期備用應急止驚藥物,及時終止癲癇發作,防止生命危險至關重要。
PIGO突變患者神經功能障礙的病因仍有待研究,目前的機制假說有兩種:一種機制假說認為是組織非特異性堿性磷酸酶(tissue non-specific alkaline phosphatase,TNAP)活性降低的結果。TNAP是一種GPI-AP,TNAP可以將維生素B6吡哆醛-5’磷酸的親水形式轉化為吡哆醛,吡哆醛是谷氨酸脫羧酶的輔因子,而谷氨酸脫羧酶是合成γ氨基丁酸(γ-aminobutyric acid,GABA)的酶,因此,GPI錨定TNAP的減少被認為會導致GABA缺乏,從而導致GPI錨定缺陷患者的癲癇發作[2,10]。吡哆醇的補充可以用來控制患有不同GPI錨生物合成疾病的患者的癲癇發作[12,13],但也僅部分患兒有效。在這一種機制中,堿性磷酸酶對細胞膜的GPI錨定活性降低,因此其向血清中的釋放增加,但不是所有的患者都有血清堿性磷酸酶水平的升高。因此有另一種機制認為,神經功能障礙的確切原因是復雜的,但可能不一定與堿性磷酸酶損傷有關。Zehavi等[4]報道的案例中,血清堿性磷酸酶水平接近正常,并發現在各種膜表達的GPI-APs中,只有CD59的表達下降,其他GPI-AP如CD16和CD24在細胞表面正常表達,這與其他報道的表達減少相反,作者認為可能是由于突變蛋白對胎兒大腦發育的有害影響,其嚴重神經系統表型可能與堿性磷酸酶樣核心區域變異的位置相關。此外,PIGO缺陷的嚴重程度與PIGO殘留活性密切相關,可以進行功能分析來確認疾病嚴重程度[2]。本研究通過文獻復習,深入該基因變異相關表型認識,后續擬進一步進行神經元過表達功能驗證,轉錄組學測序分析,深入探索其致病機制,該后續實驗研究結果,將在另一篇文章中呈現。
該基因變異目前尚無確切有效的特定治療方式,主要為對癥治療。但該基因變異癲癇發作難以控制,且容易出現癲癇持續狀態,囑托家長長期備用應急止驚藥物,及時終止癲癇發作,防止生命危險至關重要。本組1例患兒使用左乙拉西坦、丙戊酸口服,癲癇癥狀較前有所改善,其中多次出現癲癇持續狀態,予備用咪達唑侖口頰粘膜溶液應急止驚,可有效終止癲癇發作。目前在國際上,PIGO基因變異在小鼠動物模型上,已經開始了腺相關病毒載體介導的基因遞送治療,用腺相關病毒對小鼠進行同源非依賴性敲入和PIGO cDNA的染色體外表達,其癥狀及表型明顯得到改善[14]。為進一步促進該基因治療的臨床應用,未來仍需進一步研發優化的人類基因療法。
綜上所述,PIGO基因變異相關發育性癲癇性腦病較罕見,臨床表型包括全面發育遲緩、頑固性癲癇、多發畸形、腦發育不良,可伴血清堿性磷酸酶升高等。因此,在臨床上對于嬰兒早期出現生長發育遲緩、癲癇發作、肌張力低下、發育畸形、血清堿性磷酸酶水平異常等,需警惕該病,建議完善基因全外顯子測序分析以輔助診斷。該病死亡率高,容易出現癲癇持狀態,癲癇持續狀態多為早期死因,囑托家長長期備用應急止驚藥物,及時終止癲癇發作,防止生命危險至關重要。
利益沖突聲明 所有作者無利益沖突。
磷脂酰肌醇聚糖錨定生物合成O類蛋白(phosphatidylinositol glycan anchor biosynthesis class O protein,PIGO)基因編碼糖基磷脂酰肌醇-乙醇胺磷酸轉移酶3,參與糖基磷脂酰肌醇(glycosylphosphatidylinositol,GPI)錨定合成途徑的重要步驟,用于將蛋白質附著于細胞表面,維持正常神經元和胚胎發育[1]。目前報道,PIGO基因變異相關疾病存在兩種表型,一種為伴有精神發育遲滯綜合征(hyperphosphatasia with mental retardation syndrome,HPMRS/Mabry syndrome)的高磷酸酯酶癥,是一種常染色體隱性遺傳的智力障礙,其特征為面容畸形、發育遲緩和血清堿性磷酸酶持續升高(高磷酸鹽血癥)[2,3]。PIGO基因變異另一種表型為發育性癲癇性腦病,可伴胼胝體發育異常,伴或不伴輕度堿性磷酸酶增高[4]。本研究通過總結福建省兒童醫院神經內科2023年8月收治的1例患兒的臨床特點,并文獻復習,提高臨床認識。本研究經福建省兒童醫院醫學倫理委員會審核批準(2023ETKLR11006)及患兒監護人知情同意。
病例資料 患兒 男,6月齡20天,因“間斷抽搐5 h余”入院。患兒于入院前5 h于家中洗澡后出現抽搐發作,表現為意識喪失,雙眼正前方凝視,右側眼瞼頻繁眨動,口唇青紫,持續約1 h緩解,緩解后發作間歇期意識不清,間隔半小時后再發抽搐,發作形式基本同前,偶伴有不自主尖叫,抽搐時體溫38℃,就診當地醫院予苯巴比妥、地西泮止驚,期間血氧飽和度下降,最低降至78%,予氣管插管呼吸機輔助機械通氣,遂轉入我院,轉診途中仍有間斷抽搐,表現同前,間斷持續約5 h余至我院。患兒系G1P1,足月剖宮產,出生體重3.6 kg,出生史無異常,生后母乳喂養,新生兒期因“病理性黃疸”住院治療,入院前9天因“支氣管肺炎”在當地醫院住院治療。患兒生長發育較同齡兒明顯落后,目前豎頭不穩,不會翻身,不能追聲追物。4月齡開始一直行康復訓練,發育未見進步。父母健康,非近親結婚,無類似疾病家族遺傳史。入院查體:氣管插管呼吸機輔助通氣鎮靜狀態,營養不良,雙肺呼吸音粗,未聞及干濕啰音,心臟腹部查體無異常。四肢自主活動少,四肢肌張力低,肌力查體不配合。跟腱、膝腱反射可引出。腦膜刺激征陰性,病理征陰性。入院輔助檢查:血常規、血生化、血清堿性磷酸酶、腦脊液常規、生化、培養、甲功、凝血、血尿代謝篩查等均大致正常。視頻腦電圖提示醒睡各期雙側頂枕顳區棘波、棘慢波、尖形慢波、不規則慢波發放。頭顱核磁提示雙側側腦室增寬,胼胝體發育不良(圖1)。入院后予左乙拉西坦口服抗癲癇發作等對癥支持治療,逐漸撤離呼吸機,住院治療10天好轉后出院。出院間隔1個月后再發抽搐,發作形式基本同前,頻率較前增多,發作頻繁時每天數次,容易出現癲癇持續狀態,多數持續10 min以上,應用咪達唑侖口頰粘膜溶液止驚后發作可終止。后加用丙戊酸口服抗癲癇發作治療,抽搐頻率較前下降,多在感染等情況下誘發發作。后行基因全外顯子測序分析,結果顯示PIGO基因復合雜合變異:c.2607_2609del(p.Leu870del)雜合變異,變異來自母親,父親為野生型;c.3070-26A>T雜合變異,變異來自父親,母親為野生型(圖2)。根據ACMG評級、基因多態性、基因保守性、等位基因頻率等評估致病性,使用SIFT、Polyphen HDIV、Polyphen HVAR、LRT、Mutation Taster和Mutation Assessor多種軟件預測,變異均為有害,Splice AI預測c.3070-26A>T變異,預測分值0.58,遠高于常規剪接位點變異的預測閾值,該變異很可能會影響剪接,故綜合評判考慮為可能致病性復合雜合變異,結合文獻報道的該基因的兩種表型,最終該患兒診斷為PIGO基因變異相關發育性癲癇性腦病不伴輕度堿性磷酸酶增高。目前長期口服左乙拉西坦、丙戊酸抗癲癇發作藥物治療,持續隨訪中。
圖1
頭顱核磁共振提示胼胝體發育不良
圖2
PIGO基因復合雜合變異
c.2607_2609del(p.Leu870del)雜合變異,變異來自母親,父親為野生型(a,b,c);c.3070-26A>T雜合變異,變異來自父親,母親為野生型(d,e,f)
文獻復習 分別以“PIGO、PIGO mutation、發育性癲癇性腦病、癲癇性腦病”為檢索詞檢索PubMed、中國知網、萬方醫學網等數據庫(建庫至2024年6月),發現檢索符合條件的中文文獻0篇、英文文獻11篇共20例PIGO基因變異相關發育性癲癇性腦病患兒[1-11]。所有報道的患者,年齡9月齡~22歲,均出現全面性發育遲緩或智力障礙(通常為重度),其余臨床特征包括:癲癇發作(13/20)、先天性巨結腸和肛門閉鎖等腸道畸形(12/20)、指畸形(包括短指畸形和指甲發育不全)(10/20)、感音神經性聽力損失(6/20)、舞蹈病(4/20)、先天性泌尿生殖系統畸形(3/20)、先天性心臟病(3/20)和食管閉鎖(2/20)。顱縫早閉和椎體異常各有1例。11/20個體存在面部畸形,常見的有寬瞼裂、高鼻梁、腭裂等。15/20個體存在高磷酸鹽血癥(即血清堿性磷酸酶水平升高)。1例病程中并發脫水、低血糖伴功能性高胰島素血癥、皮質醇減退癥以及橫紋肌溶解癥。神經影像學檢查結果包括腦白質病變(5/20)、腦萎縮伴腦室腫大(5/20)、小腦萎縮(4/20)、胼胝體薄(3/20)、基底神經節或腦干彌散受限(3/20)以及視神經發育不良(1/20)。20例PIGO基因變異位點及對應臨床表型特點總結見表1。
20例患兒中13例出現癲癇發作,癲癇發作起病年齡為4日齡~1歲11月齡,癲癇發作形式包括局灶性發作、強直陣攣發作、全面性強直發作、全面性強直陣攣發作,聯合應用2~4種抗癲癇發作藥物,包括苯巴比妥、拉莫三嗪、托吡酯、左乙拉西坦、丙戊酸、氯巴占、司替戊醇、維生素B6、吡哆醇等,其中2例癲癇發作控制,11例未控制。其中1例使用吡哆醇治療有效,每日口服400 mg吡哆醇(20 mg/kg)一周后,觀察到癲癇發作完全停止,發作間期腦電圖改善,吡哆醇給藥中斷容易引起癲癇再次發作,需長期口服維持。1例添加丙戊酸治療,發作控制。最終6例死亡(6/20,30%),死亡年齡為9月齡~2歲8月齡,其中2例c.1109A>G (p.N370S)、c.2497_2498del (p.A834Cfs)復合雜合變異患兒分別死于17月齡、20月齡,1例c.2869C>T(p.L957P)、c.3069+5G>A復合雜合變異患兒死于22月齡,1例c.765G>A (p.M255I)純合變異患兒死于2歲8月齡,1例c.1352T>G (p.Met451Arg)、c.1392delinsGA (p.Ile464Mfs*42)復合雜合變異患兒死于9月齡,1例c.389C>A (p.Thr130Asn)、c.1288C>T (p.Gln430*) 復合雜合變異患兒死于1歲。6例中2例因驚厥持續狀態死亡。
討論 遺傳性GPI缺乏癥(inherited glycosylphosphatidylinositol deficiencies,IGDs)也稱為IGDs,至少有26個基因參與GPI錨定蛋白的生物合成和運輸,目前已有14個基因突變被確定為常染色體隱性遺傳的IGDs的原因:PIGQ、PIGY、PIGC、PIGL、PIGW、PIGM、PIGV、PIGN、PIGO、PIGG、PIGT、PGAP1、PGAP2和PGAP3。其中,PIGV和PIGO突變是最常見的原因[1,2]。IGDs導致GPI錨定蛋白(GPI-anchored proteins,GPI-Aps)表達降低,影響神經元功能和胚胎發育,參與疾病發生發展。
IGDs的臨床表現多種多樣,其特征包括智力障礙、頑固性癲癇、畸形特征和器官異常,臨床表型嚴重程度也多種多樣。由于IGDs是罕見疾病,表型與基因型的相關性尚未得到很好的定義[2]。本組患兒1例,以全面性發育遲緩、癲癇發作不伴有血清磷酸酶升高為主要表現,與文獻中報道的表型符合。臨床上需要注意的是,該基因變異相關癲癇,容易出現癲癇持續狀態,癲癇持續狀態多為早期死因,囑托家長長期備用應急止驚藥物,及時終止癲癇發作,防止生命危險至關重要。
PIGO突變患者神經功能障礙的病因仍有待研究,目前的機制假說有兩種:一種機制假說認為是組織非特異性堿性磷酸酶(tissue non-specific alkaline phosphatase,TNAP)活性降低的結果。TNAP是一種GPI-AP,TNAP可以將維生素B6吡哆醛-5’磷酸的親水形式轉化為吡哆醛,吡哆醛是谷氨酸脫羧酶的輔因子,而谷氨酸脫羧酶是合成γ氨基丁酸(γ-aminobutyric acid,GABA)的酶,因此,GPI錨定TNAP的減少被認為會導致GABA缺乏,從而導致GPI錨定缺陷患者的癲癇發作[2,10]。吡哆醇的補充可以用來控制患有不同GPI錨生物合成疾病的患者的癲癇發作[12,13],但也僅部分患兒有效。在這一種機制中,堿性磷酸酶對細胞膜的GPI錨定活性降低,因此其向血清中的釋放增加,但不是所有的患者都有血清堿性磷酸酶水平的升高。因此有另一種機制認為,神經功能障礙的確切原因是復雜的,但可能不一定與堿性磷酸酶損傷有關。Zehavi等[4]報道的案例中,血清堿性磷酸酶水平接近正常,并發現在各種膜表達的GPI-APs中,只有CD59的表達下降,其他GPI-AP如CD16和CD24在細胞表面正常表達,這與其他報道的表達減少相反,作者認為可能是由于突變蛋白對胎兒大腦發育的有害影響,其嚴重神經系統表型可能與堿性磷酸酶樣核心區域變異的位置相關。此外,PIGO缺陷的嚴重程度與PIGO殘留活性密切相關,可以進行功能分析來確認疾病嚴重程度[2]。本研究通過文獻復習,深入該基因變異相關表型認識,后續擬進一步進行神經元過表達功能驗證,轉錄組學測序分析,深入探索其致病機制,該后續實驗研究結果,將在另一篇文章中呈現。
該基因變異目前尚無確切有效的特定治療方式,主要為對癥治療。但該基因變異癲癇發作難以控制,且容易出現癲癇持續狀態,囑托家長長期備用應急止驚藥物,及時終止癲癇發作,防止生命危險至關重要。本組1例患兒使用左乙拉西坦、丙戊酸口服,癲癇癥狀較前有所改善,其中多次出現癲癇持續狀態,予備用咪達唑侖口頰粘膜溶液應急止驚,可有效終止癲癇發作。目前在國際上,PIGO基因變異在小鼠動物模型上,已經開始了腺相關病毒載體介導的基因遞送治療,用腺相關病毒對小鼠進行同源非依賴性敲入和PIGO cDNA的染色體外表達,其癥狀及表型明顯得到改善[14]。為進一步促進該基因治療的臨床應用,未來仍需進一步研發優化的人類基因療法。
綜上所述,PIGO基因變異相關發育性癲癇性腦病較罕見,臨床表型包括全面發育遲緩、頑固性癲癇、多發畸形、腦發育不良,可伴血清堿性磷酸酶升高等。因此,在臨床上對于嬰兒早期出現生長發育遲緩、癲癇發作、肌張力低下、發育畸形、血清堿性磷酸酶水平異常等,需警惕該病,建議完善基因全外顯子測序分析以輔助診斷。該病死亡率高,容易出現癲癇持狀態,癲癇持續狀態多為早期死因,囑托家長長期備用應急止驚藥物,及時終止癲癇發作,防止生命危險至關重要。
利益沖突聲明 所有作者無利益沖突。