

引用本文: 珊措吉, 李君麗, 陳茂. 過氧化物酶體增殖物激活受體 γ 共激活因子 1α 抑制主動脈瓣瓣膜間質細胞活化的作用及機制研究. 華西醫學, 2024, 39(9): 1387-1397. doi: 10.7507/1002-0179.202402173 復制
版權信息: ?四川大學華西醫院華西期刊社《華西醫學》版權所有,未經授權不得轉載、改編
主動脈瓣狹窄(aortic stenosis, AS)是先天性因素如瓣葉發育畸形或后天性因素如退行性變、風濕性變誘發的主動脈瓣進行性重塑出現纖維化、鈣化,而造成主動脈瓣葉增厚粘連、瓣口狹窄、左心室流出道受阻進而導致左心室不良重塑的疾病[1]。AS 是最常見的心臟瓣膜病,發病率及病死率高,但目前尚無有效的早期干預措施及治療藥物。因此,對 AS 的發病機制進行深入研究,并推動潛在藥物治療靶點的發現顯得尤為重要。AS 誘因多樣、發病機制復雜。其中,主動脈瓣的主要細胞成分主動脈瓣瓣膜間質細胞(aortic valve interstitial cells, AVIC)發生活化并向成骨分化被認為是 AS 的關鍵環節[2]。但目前 AVIC 的活化機制尚未闡明。臨床前和人體研究數據表明,各種來源的活性氧的不良積累介導了 AS 起始階段的浸潤性脂質的氧化和炎癥反應,進而誘導 AVIC 出現肌成纖維細胞樣表型活化,且氧化應激水平升高程度與 AS 的嚴重程度相關[3]。據報道,脂質氧化及其介導的細胞表型轉換可受過氧化物酶體增殖物激活受體 γ 共激活因子 1α(peroxisome proliferator-activated receptor gamma coactivator 1α, PGC-1α)的調控[4]。PGC-1α 是控制多種核編碼的線粒體基因的關鍵分子,可通過激活多種轉錄因子調控驅動線粒體呼吸鏈和脂肪酸氧化酶基因的表達,促使線粒體數目增加、線粒體呼吸能力增強[5]。既往研究表明,PGC-1α 可作為氧化應激的關鍵抑制因子降低活性氧生成、減少氧化應激及炎癥反應[6]。更重要的是,PGC-1α 可通過調控核呼吸因子而調節線粒體轉錄因子 A、B1、B2 的表達繼而調節線粒體基質蛋白的表達發揮活性氧脫毒作用。PGC-1α 包含參與 RNA 調控的蛋白質結構域如絲氨酸/精氨酸和 RNA 識別基序,可直接調控 RNA 的表達,而 AVIC 活化進程中 PGC-1α 是否通過作用于特殊 RNA 而發揮作用仍尚未明確。既往 PGC-1α 免疫共沉淀后的轉錄組測序結果顯示,其可能與鈣調蛋白依賴性蛋白激酶 1δ(calcium/calmodulin-dependent protein kinase 1δ, CAMK1δ)、生物鐘基因周期蛋白 1(period 1, PER1)、經典瞬時受體電位通道 5(transient receptor potential canonical 5, TRPC5)、叉頭框蛋白 O1(fork-head box protein O1, FOXO1)等存在相互作用[7]。目前,PGC-1α 在 AS 關鍵環節即 AVIC 表型轉變中的作用及機制尚未可知。因此,本研究旨在探討 AVIC 活化進程中 PGC-1α 的表達水平改變情況、作用特點及機制途徑。
1 材料與方法
1.1 實驗試劑
高糖基礎培養基、減血清培養基、0.25% 胰蛋白酶、Lipofectamine RNAiMAX 試劑、Ⅱ型膠原酶、胎牛血清及總 RNA 抽提試劑盒 TRIzolTM均購自美國 Thermo Fisher Scientific 公司;轉化生長因子 β1(transforming growth factor β1, TGF-β1)購自北京義翹神州科技股份有限公司;聚偏二氟乙烯膜、4’, 6-二脒基-2-苯基吲哚(4’, 6-diamidino-2-phenylindole, DAPI)染液購自美國 Merck 公司;逆轉錄酶購自日本 Toyobo 公司;實時定量聚合酶鏈反應(real-time quantitative polymerase chain reaction, RT-qPCR)熒光染料預混液購自美國 BIO-RAD 公司;Triton?X-100 和 4% 多聚甲醛固定液購自中國 Biosharp 公司;活性氧檢測試劑盒(S0033)、細胞裂解液購自上海碧云天生物技術有限公司;小干擾 RNA(small interfering RNA, siRNA)由中國吉瑪基因公司合成;辣根酶標記山羊抗兔/小鼠免疫球蛋白 G 購自中國中杉金橋生物公司。抗體信息:Alexa Fluor? 488 Conjugate 熒光二抗和兔抗磷酸化哺乳動物雷帕霉素靶蛋白(phospho-mammalian target of rapamycin, p-mTOR)抗體購自美國 Cell Signaling Technology 公司;兔抗 β-微管蛋白(β-tubulin)和兔抗 β-肌動蛋白(β-actin)抗體購自武漢愛博泰克生物科技有限公司;兔 PGC-1α 抗體購自美國 Novus 公司;兔抗骨膜蛋白抗體、兔抗Ⅰ型膠原蛋白抗體、鼠抗甘油醛-3-磷酸脫氫酶(glyceraldehyde-3-phosphate dehydrogenase, GAPDH)抗體、鼠抗 α-平滑肌肌動蛋白(α-smooth muscle actin, α-SMA)抗體和兔抗 CAMK1δ 抗體均購自英國 Abcam 公司。
1.2 細胞培養及 AVIC 活化模型構建
用以提取 AVIC 的 6 周齡、體重 200~250 g 的雄性 SD 大鼠購自成都達碩實驗動物有限公司(動物操作獲四川大學華西醫院動物倫理委員會批準,批件號:20230920002)。該研究所使用的主動脈夾層患者來源的主動脈瓣已通過四川大學華西醫院生物醫學倫理委員會的批準(批準號:201793)。本研究符合赫爾辛基宣言中所述的人體組織使用原則。AVIC 接種于含有 20% 胎牛血清、1% 青霉素-鏈霉素的高糖培養基中,5% 二氧化碳孵箱中培養,待細胞融合度達 95% 左右進行傳代,并將血清減半。
作為誘導成纖維細胞活化的經典刺激,細胞因子 TGF-β1 目前也被廣泛用以刺激 AVIC 以誘導其活化[8],本實驗采用 30 ng/mL 的 TGF-β1 重組蛋白構建大鼠 AVIC 活化模型。實驗分組(n=4):① 未加 TGF-β1 的空白對照組;② 加 TGF-β1 刺激 24 h 組;③ 加 TGF-β1 刺激 48 h 組;④ 加 TGF-β1 刺激 72 h 組。通過檢測纖維化標志物結締組織生長因子(connective tissue growth factor, CTGF)[9]、骨膜蛋白[10]或Ⅰ型膠原蛋白[11]、α-SMA[12]的表達水平變化評估模型是否構建成功。
1.3 細胞轉染及分組
1.3.1 體外 AVIC 過表達重組腺病毒轉染及抑制劑干預
為探討 PGC-1α 在 AVIC 活化進程中的作用,本研究通過腺病毒轉染的方法外源特異性過表達/使用過氧化物酶體增殖物激活受體 γ(peroxisome proliferator-activated receptor γ, PPARγ)拮抗劑 GW9662 抑制大鼠 AVIC 中 PGC-1α 的表達。
本實驗均以 12 孔板細胞培養板 1 mL 體系為例進行干預。因活化標志物均在 TGF-β1 刺激 48 h 時開始顯著上調,后續體外實驗均采用 TGF-β1 誘導 AVIC 48 h 進行模型構建。轉染 PGC-1α 過表達腺病毒(ADPGC-1α)外源性上調 PGC-1α 的表達,預實驗結果顯示,與空白對照 ADC 組相比,以最佳感染復數為 300 時攜帶的 PGC-1α 過表達序列的腺病毒(ADPGC-1α 組)轉染能使 AVIC 中 PGC-1α 的蛋白和基因表達水平均顯著增加,并以該方案下的 PGC-1α 過表達效率用于后續實驗。大鼠及人源 AVIC 轉染過表達腺病毒的實驗分組(n=3):① 空白對照 ADC 組;② ADC+TGF-β1 組;③ ADPGC-1α 組;④ ADPGC-1α+TGF-β1 組。隨后使用 PPARγ 拮抗劑 GW9662 抑制大鼠 AVIC 中 PGC-1α 的生物活性[13],實驗分 4 組(n=3):① 空白對照二甲基亞砜(dimethyl sulfoxide, DMSO)組;② DMSO+TGF-β1 組;③ GW9662 組;④ GW9662+TGF-β1 組。隨后以 RT-qPCR、蛋白質印跡法、細胞免疫熒光法觀察 AVIC 活化程度以及活性氧水平的改變情況。
1.3.2 體外 AVIC 小干擾 RNA 轉染
為進一步探索 PGC-1α 調節 AVIC 活化的下游分子和下游信號通路,結合文獻報道,以 siRNA 特異性沉默大鼠 AVIC 中 CAMK1δ 的表達,分為 4 組(n=3):① 空白對照 SiNC 組;② SiNC+TGF-β1 組;③ SiCAMK1δ 組;④ SiCAMK1δ+TGF-β1 組。隨后以 RT-qPCR、蛋白質印跡法觀察 AVIC 活化程度的改變情況。
1.4 流式細胞術
本研究發現 PGC-1α 可通過下調 CAMK1δ 發揮作用,而 CAMK1δ 的下調與活性氧生成減少密切相關,那么在 AVIC 活化進程中,PGC-1α 是否可通過抑制活性氧生成而發揮作用尚未可知。而活性氧與多種細胞信號通路的激活密切相關,因此,本研究篩選了 PGC-1α 下游可能作用的信號通路激活情況,如活性氧可作用于的哺乳動物雷帕霉素靶蛋白(mechanistic target of rapamycin, mTOR)及可作用于活性氧生成的低氧誘導因子 1α(hypoxia-inducible factor-1α, HIF-1α)等。隨后以流式細胞實驗觀察大鼠 AVIC 中活性氧水平的改變情況。
通過 BD FACSCanto?流式分析儀(美國 BD 公司)檢測熒光標記的細胞內活性氧及 PGC-1α 分子。將大鼠 AVIC 傳代細胞鋪于 12 孔細胞培養板內待細胞融合度達 70%,根據實驗需求處理細胞。待收樣時棄去原培養基,分別加入稀釋的熒光探針加載 2’,7’-二氯熒光黃雙乙酸鹽試劑盒(1∶
1.5 細胞免疫熒光染色實驗
根據實驗設計,待達到細胞實驗時間節點,棄去原培養基,用磷酸鹽緩沖液(phosphate buffer saline, PBS)漂洗 3 次,室溫下加入 4% 多聚甲醛固定液固定 15~20 min 后再用 PBS 漂洗 3 次,用膠頭滴管吸凈 PBS 液,加入含有 0.5% 細胞通透劑 Triton?X-100 和 1% 牛血清蛋白的混合液,室溫封閉穿孔 60 min 后用 PBS 漂洗 3 次,3 min/次。隨后加入稀釋后的 α-SMA 一抗(1∶200),4℃孵育過夜處理。第 2 天回收一抗后用 PBS 漂洗 3 次,3 min/次。加入免疫熒光二抗后室溫下避光孵育 1 h 后用 PBS 漂洗 3 次,3 min/次。滴加 DAPI(用 PBS 稀釋為 1∶
1.6 RT-qPCR 檢測基因的表達變化
通過 RT-qPCR 對基因轉錄水平進行檢測。將 TRIzol 裂解液加入 12 孔板細胞培養板中裂解細胞,通過氯仿法提取總 RNA。通過與逆轉錄酶孵育,根據表1 配制逆轉錄反應體系,本研究中使用的引物序列見表2,將總 RNA 逆轉錄為單鏈互補 DNA。根據制造商的說明,使用試劑盒進行 RT-qPCR 檢測。在 CFX Connect 機器(美國 BIO-RAD 公司)上檢測到信號。GAPDH 或 β-actin 作為內參基因,即相對基因表達歸一化為 GAPDH 表達,進行定量分析。
1.7 蛋白質印跡法檢測蛋白表達
根據實驗設計待細胞實驗完成后,將培養的細胞直接在放射免疫沉淀試驗緩沖液中裂解,冰上裂解 15 min,4℃,
1.8 統計學方法
使用 GraphPad Prism 8.0 軟件進行統計分析,實驗結果以均數±標準差表示。兩組間比較使用兩獨立樣本 t 檢驗,多組間比較使用單因素方差分析,組間兩兩比較使用SNK-q檢驗。雙側檢驗水準 α=0.05。
2 結果
2.1 PGC-1α 表達水平在活化的大鼠 AVIC 中下降
2.1.1 TGF-β1 誘導的大鼠 AVIC 活化模型構建成功
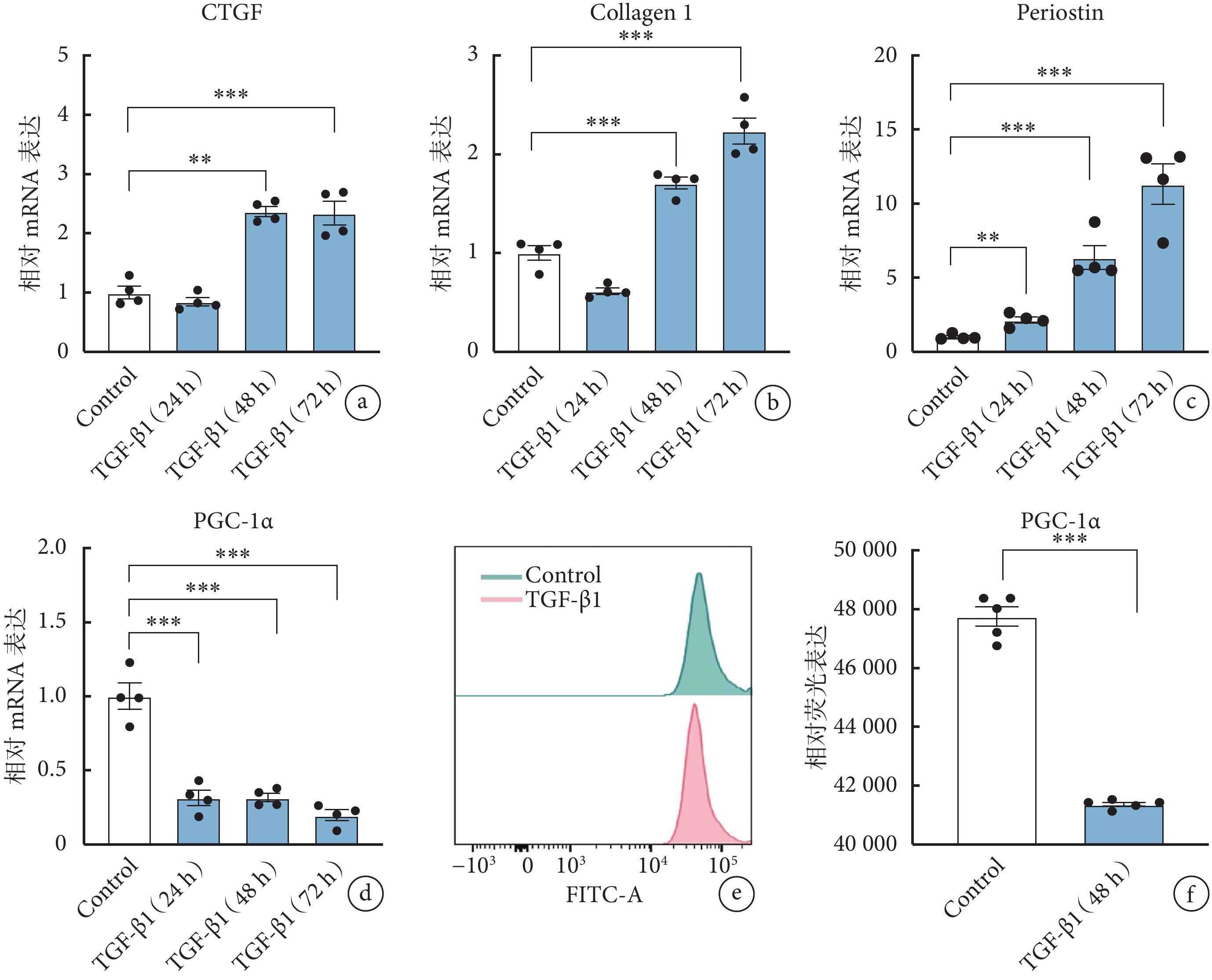
實驗結果表明,與空白對照組相比,活化標志物骨膜蛋白的 mRNA 表達水平在 TGF-β1 刺激大鼠 AVIC 24、48、72 h 后呈梯度增加現象,CTGF 及Ⅰ型膠原蛋白的 mRNA 表達水平亦在 TGF-β1 作用 48、72 h 后表達增加(圖1a~1c),表明 TGF-β1 誘導的大鼠 AVIC 活化模型構建成功。
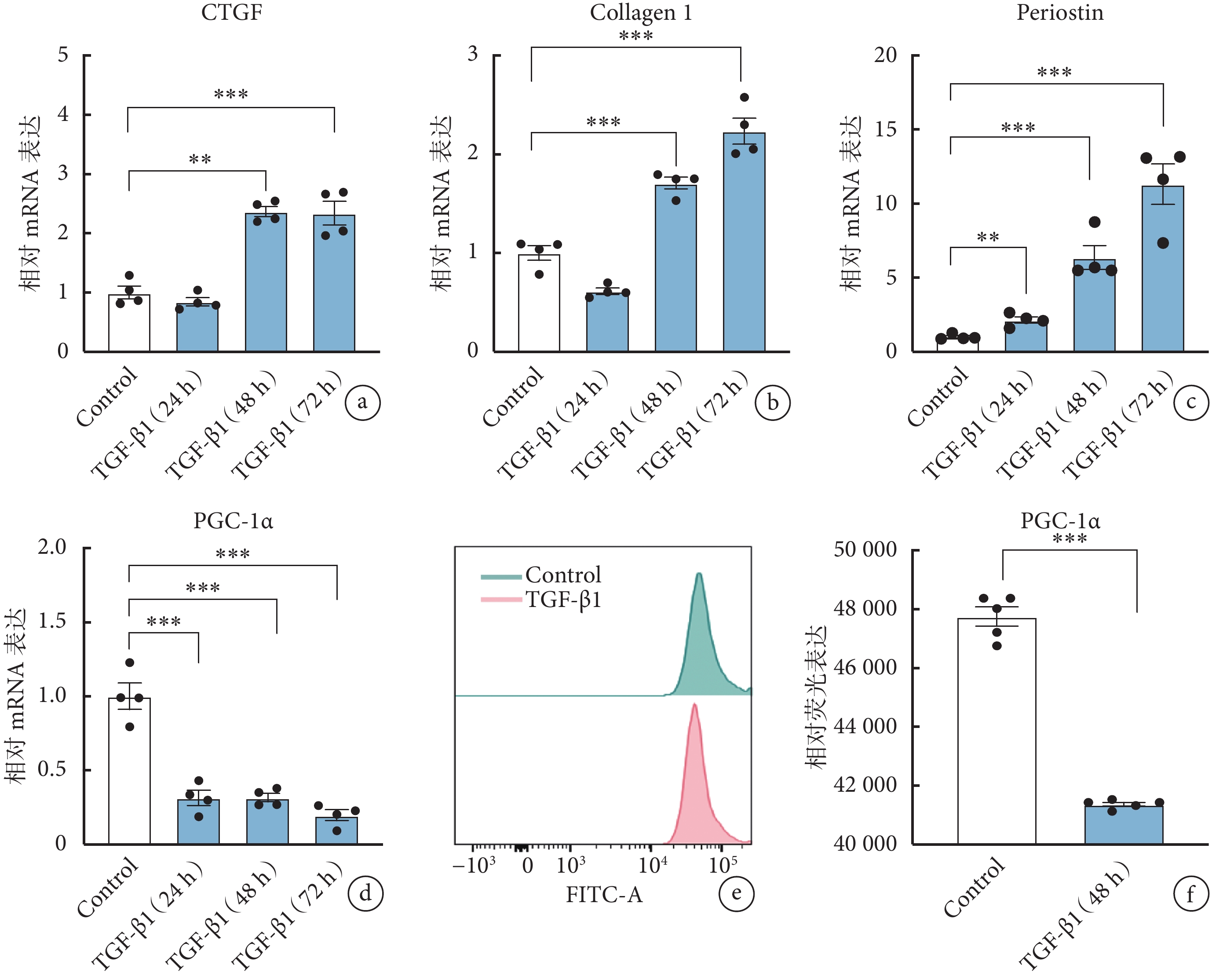 圖1
PGC-1α 在活化的 AVIC 中表達降低
圖1
PGC-1α 在活化的 AVIC 中表達降低
a~d. 大鼠 AVIC 在 TGF-β1 刺激 24、48、72 h 后,各纖維化標志物及 PGC-1α 的 mRNA 表達水平(
2.1.2 PGC-1α 在 TGF-β1 誘導活化的大鼠 AVIC 中表達下調
與空白對照組(1.00±0.18)相比,TGF-β1 刺激大鼠 AVIC 24、48、72 h 后,PGC-1α 的 mRNA 表達水平均出現明顯下調(24 h:0.31±0.10;48 h:0.32±0.06;72 h:0.20±0.07),且差異有統計學意義(P<0.05,圖1d)。流式細胞術結果顯示,TGF-β1 刺激 AVIC 48 h 后,PGC-1α 的蛋白表達水平亦顯著下調(圖1e、1f)。
2.2 PGC-1α 可緩解大鼠 AVIC 活化程度
2.2.1 過表達 PGC-1α,AVIC 活化程度減輕
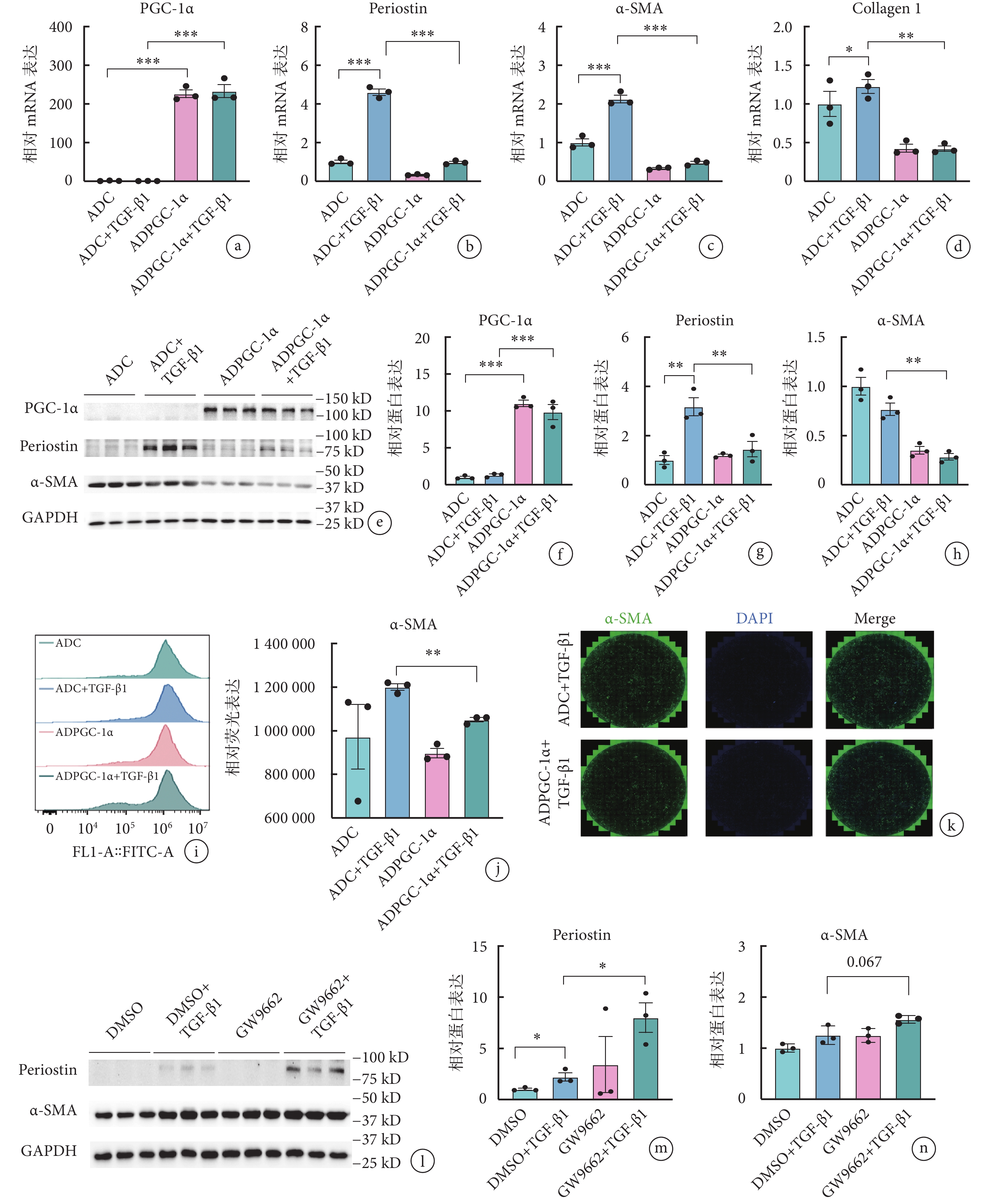
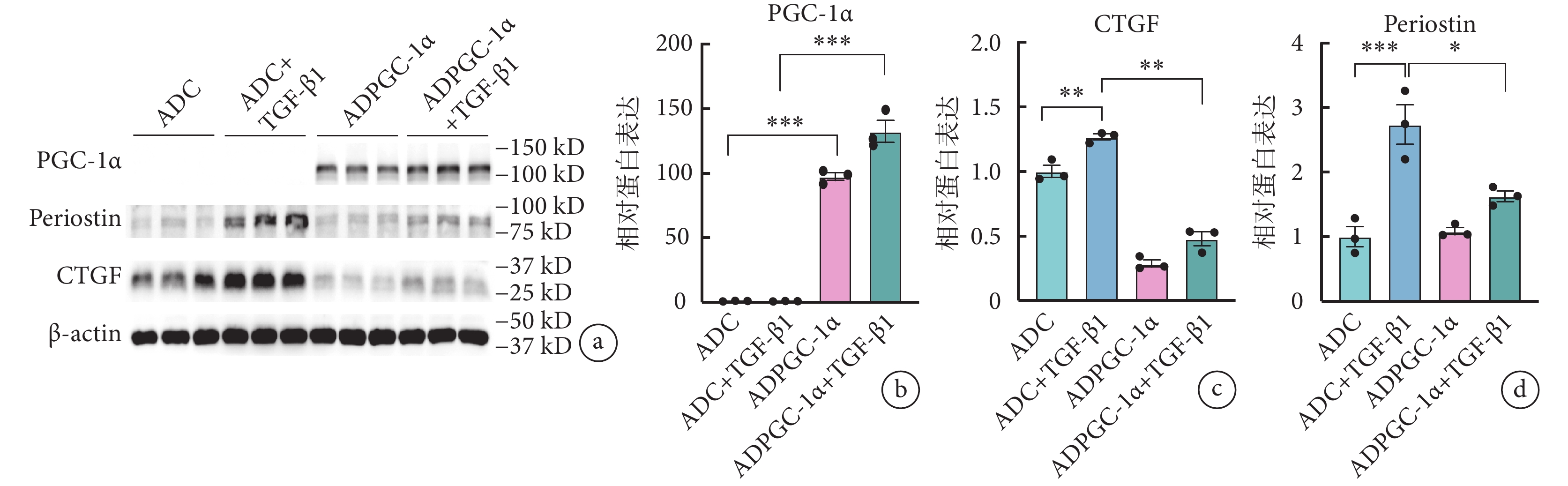
與對照組(ADC+TGF-β1 組)相比,過表達 PGC-1α 后(ADPGC-1α+TGF-β1 組),伴隨 PGC-1α 的表達明顯上調,TGF-β1 誘導的纖維化標志物骨膜蛋白、α-SMA 的基因和蛋白表達均明顯降低(骨膜蛋白:3.17±0.64 vs. 1.45±0.54,P<0.05;α-SMA:0.77±0.11 vs. 0.28±0.06,P<0.05),見圖2a~2h。流式細胞術和免疫熒光結果均驗證了高表達 PGC-1α 后,TGF-β1 誘導的 α-SMA 表達水平出現明顯降低(圖2i~2k)。
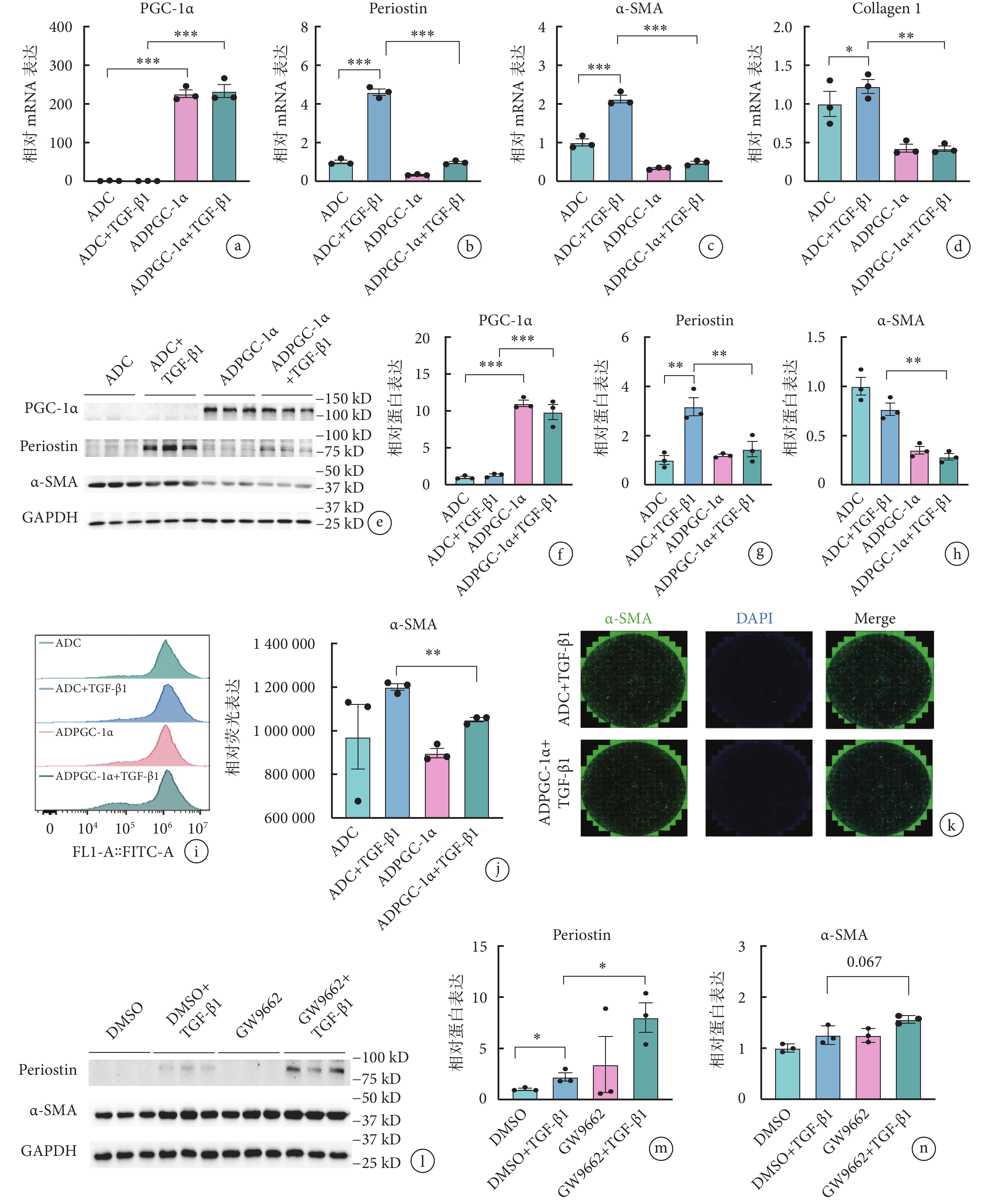 圖2
PGC-1α 可緩解大鼠 AVIC 活化程度
圖2
PGC-1α 可緩解大鼠 AVIC 活化程度
a~d. 轉染 ADPGC-1α 前后各組大鼠 AVIC 的 PGC-1α 及骨膜蛋白、α-SMA、Ⅰ型膠原蛋白的 mRNA 表達水平(
2.2.2 抑制 PGC-1α 活性,AVIC 活化程度增加
進一步反向驗證 PGC-1α 在 AVIC 活化進程中的作用發現,GW9662 抑制 PGC-1α 活性可進一步上調 TGF-β1 所誘導的骨膜蛋白表達水平,差異有統計學意義(2.20±0.68 vs. 7.99±2.50,P<0.05),同時 α-SMA 表達也呈現出上調趨勢(1.25±0.18 vs. 1.56±0.07,P=0.067)。見圖2l~2n。
2.3 PGC-1α 通過下調 CAMK1δ 的表達發揮作用
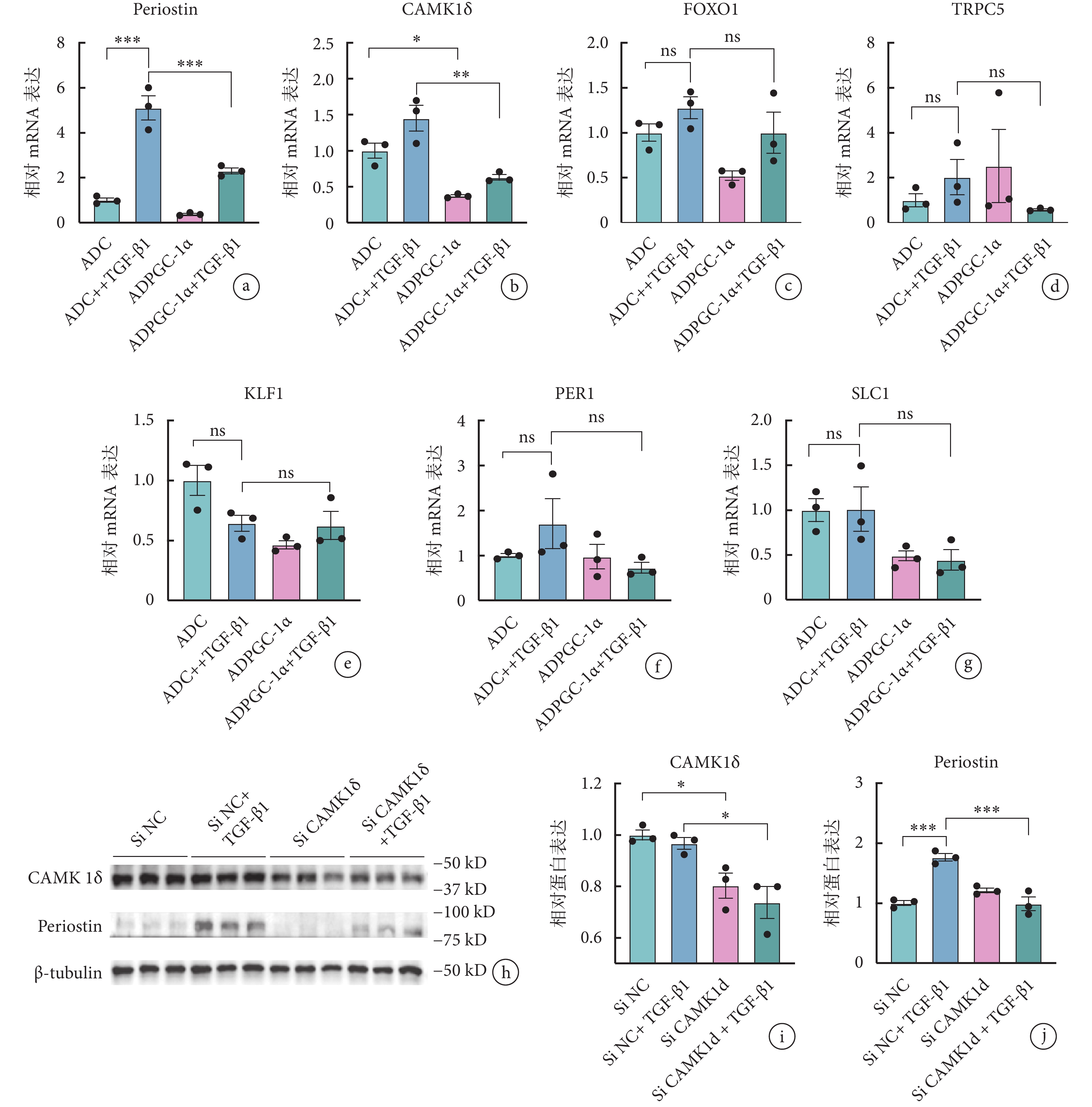
過表達 PGC-1α 表達水平后進行下游篩選,鑒定到 PGC-1α 可能通過下調 CAMK1δ 的表達發揮作用(0.97±0.04 vs. 0.74±0.11,P<0.05)(圖3a~3g)。隨后在 TGF-β1 誘導下,使用 Si CAMK1δ 特異性沉默其表達水平后觀察 AVIC 活化程度改變情況,結果顯示,沉默 CAMK1δ 表達下調了活化標志物骨膜蛋白的表達(1.76±0.11 vs. 0.99±0.20,P<0.05),表明 AVIC 活化程度緩解(圖3h~3j)。
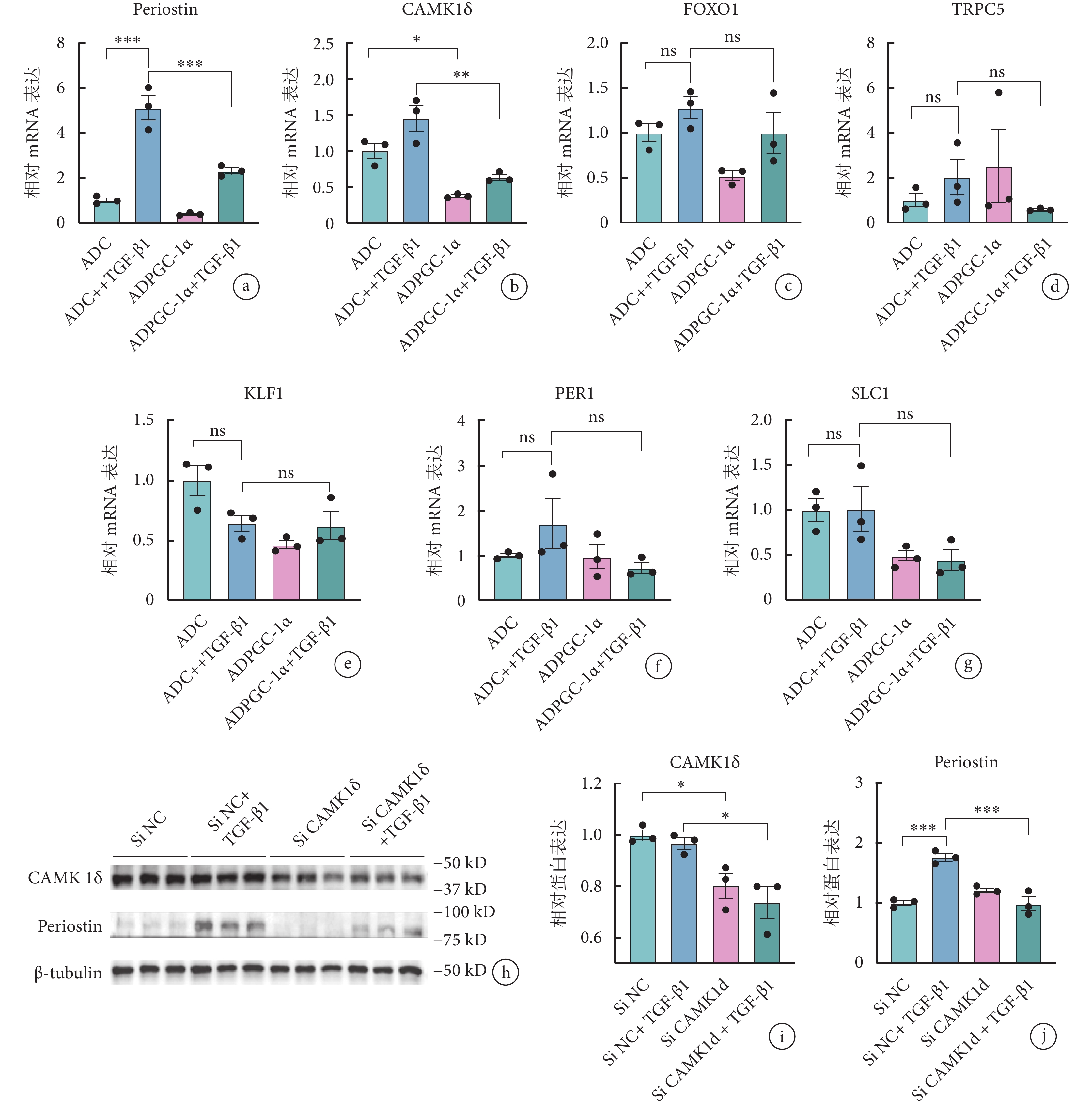 圖3
PGC-1α 可通過下調 CAMK1δ 表達水平發揮作用
圖3
PGC-1α 可通過下調 CAMK1δ 表達水平發揮作用
a~g. 轉染 ADPGC-1α 前后各組大鼠 AVIC 的骨膜蛋白及
2.4 PGC-1α 抑制活性氧生成而發揮作用
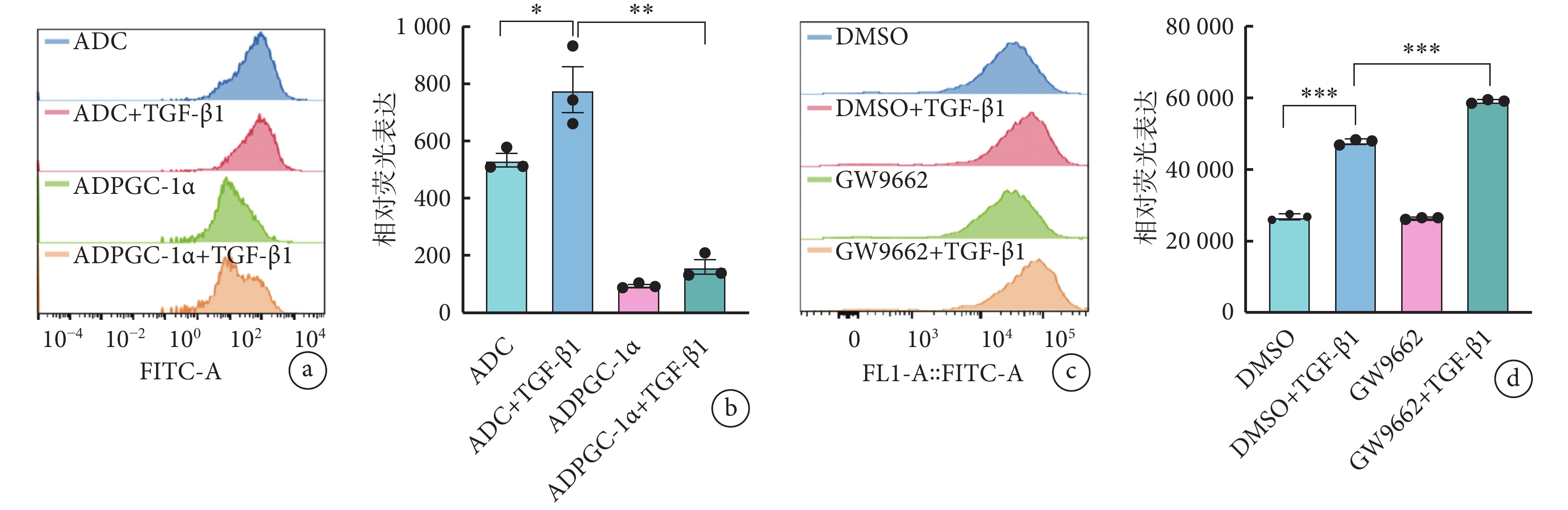
流式細胞學結果顯示,TGF-β1 作用下,與對照組(ADC+TGF-β1 組)相比,ADPGC-1α+TGF-β1 組活性氧活性降低且差異有統計學意義(778.3±139.4 vs. 159.3±43.2,P<0.05)(圖4a、4b)。同時,功能抑制實驗結果顯示,TGF-β1 刺激下,與對照組(DMSO+TGF-β1 組)相比,GW9662 的使用(GW9662+TGF-β1 組)進一步增加了 TGF-β1 誘導的 AVIC 活化進程中的活性氧水平(圖4c、4d)。
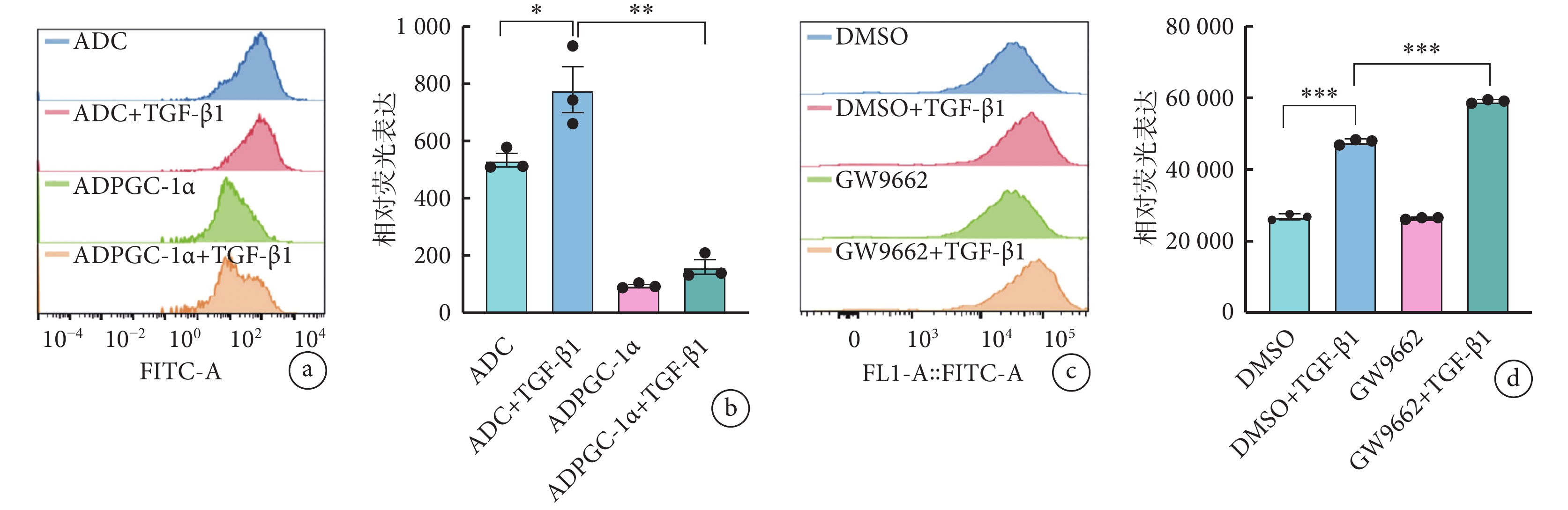 圖4
PGC-1α 抑制活性氧生成而發揮作用
圖4
PGC-1α 抑制活性氧生成而發揮作用
a、b. 存在或不存在 TGF-β1 刺激的情況下,AVIC 轉染 ADPGC-1α 后活性氧的流式細胞術結果(
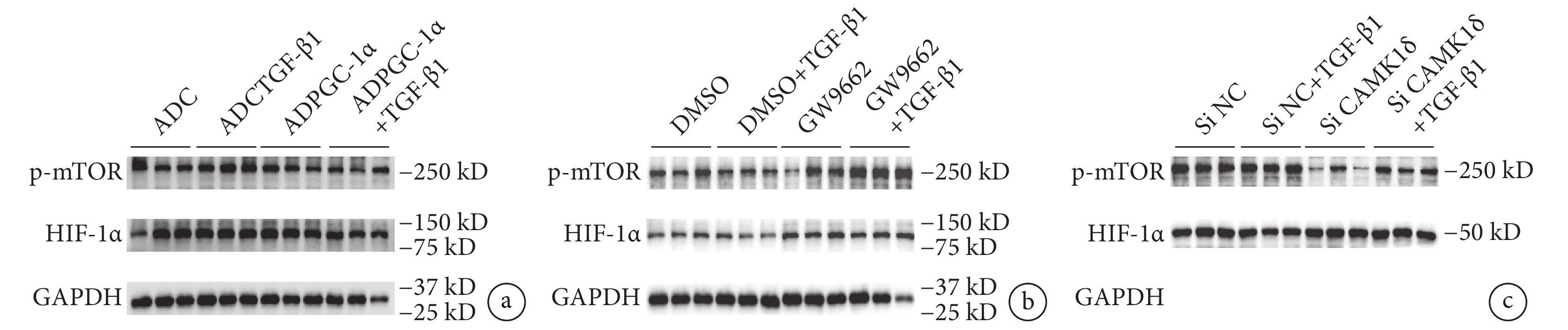
2.5 PGC-1α 負調控 p-mTOR 通路激活而發揮作用
蛋白質印跡法結果顯示,TGF-β1 刺激下,與對照組(ADC+TGF-β1)相比,ADPGC-1α+TGF-β1 組 p-mTOR 的表達明顯下調(圖5a);相反,使用 GW9662 抑制 PGC-1α 活性后,p-mTOR 通路分子表達上調(圖5b);HIF-1α 則無明顯變化。
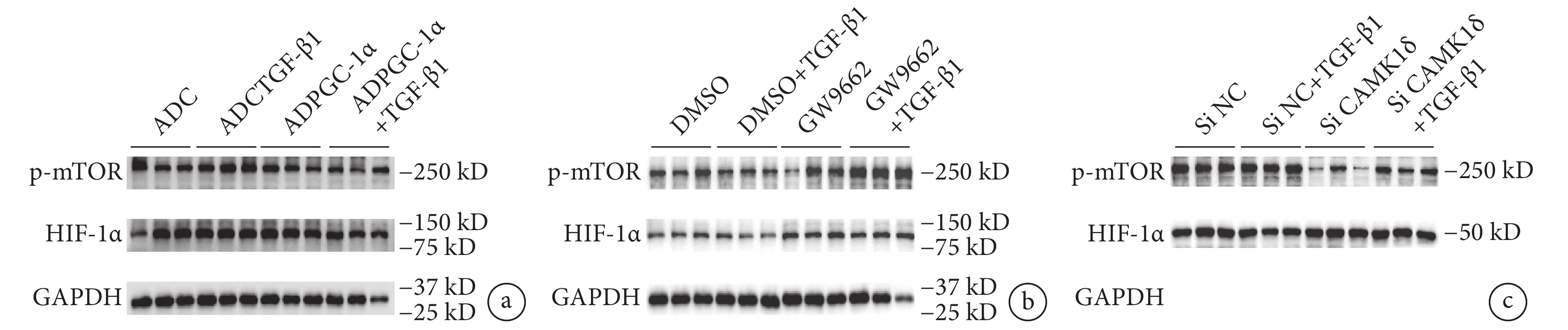 圖5
PGC-1α 可負調控 p-mTOR 通路激活而發揮作用
圖5
PGC-1α 可負調控 p-mTOR 通路激活而發揮作用
a. 存在或不存在 TGF-β1 刺激的情況下,AVIC 轉染 ADPGC-1α 后通路蛋白分子 p-mTOR、HIF-1α 的蛋白水平(
鑒于 PGC-1α 可下調 CAMK1δ 表達,而文獻報道 CAMK1δ 表達的下降與活性氧生成減少相關,隨后,我們觀察了特異性沉默 CAMK1δ 后 mTOR 通路的激活情況,結果顯示,下調 CAMK1δ 表達水平后 mTOR 通路激活程度減輕(圖5c)。
2.6 過表達 PGC-1α 緩解 TGF-β1 誘導的人源的 AVIC 活化程度
基于上述結果,通過人來源的 AVIC 進行驗證 PGC-1α 發揮作用的物種保守性。蛋白質印跡實驗結果顯示,與對照組(ADC+TGF-β1 組)相比,ADPGC-1α+TGF-β1 組活化標志物骨膜蛋白、CTGF 蛋白水平表達均明顯降低(骨膜蛋白:2.73±0.53 vs. 1.63±0.14,P<0.05;CTGF:1.27±0.04 vs. 0.48±0.09,P<0.05),差異有統計學意義(圖6)。
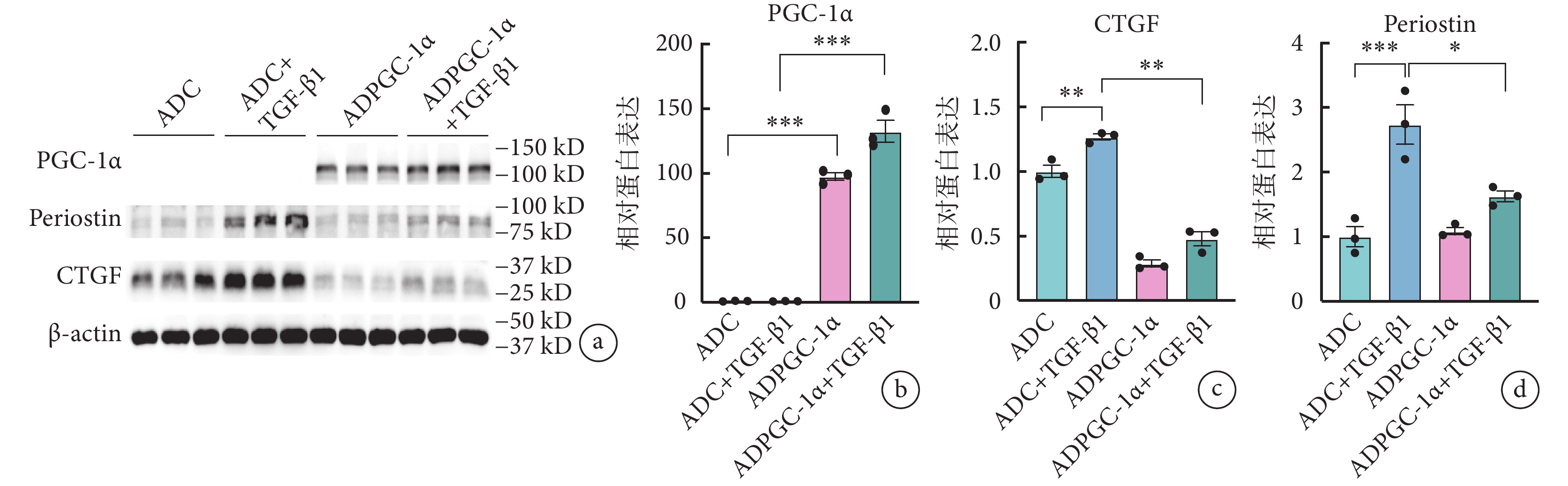 圖6
過表達 PGC-1α 可緩解 TGF-β1 誘導的人源的 AVIC 活化程度
圖6
過表達 PGC-1α 可緩解 TGF-β1 誘導的人源的 AVIC 活化程度
a~d. 存在或不存在 TGF-β1 刺激的情況下,人源 AVIC 轉染 ADPGC-1α 后骨膜蛋白、α-SMA 的蛋白表達水平(
3 討論
AS 主要病理表現為主動脈瓣葉體部纖維化鈣化增厚、主動脈瓣環狹窄、左心室的機械應力增加引起嚴重并發癥,目前尚無有效緩解藥物[14]。經導管主動脈瓣置換術是重度 AS 的成熟治療手段,但術后傳導阻滯、急性腎損傷、卒中等并發癥及瓣膜衰敗等后續不良后果的出現,使 AS 發病機制的探索及藥物治療靶點的發現變得尤為重要。
AS 誘因多樣、發病機制復雜,分子和細胞過程未被清楚表征。現有研究認為,瓣膜內皮功能受損、脂質浸潤沉積、炎癥細胞滲出、免疫炎癥反應以及在此基礎上發生的瓣膜纖維化和/或鈣化是其主要機制[15]。在此過程中 AVIC 的表型轉變包括纖維性活化及向成骨分化(即鈣化)是 AS 的關鍵環節。盡管所有終末期 AS 瓣膜都表現為鈣化,但纖維化在 AS 中始終起著重要作用[16]。因此,本研究選擇 AVIC 作為靶細胞,研究 AS 早期細胞表型轉換(即 AVIC 活化)的生物學機制,以期為輕度 AS 患者進展為重度 AS 患者的中間過程提供潛在阻遏靶點。
如前所述,一項使用硫代巴比妥酸評估全身脂質過氧化和 2, 4-二硝基苯肼評估血漿蛋白氧化修飾的隊列研究表明,氧化應激水平升高與 AS 的嚴重程度相關[17]。在分離培養的豬 AVIC 中,TGF-β1 刺激可通過激活促分裂素原活化蛋白激酶信號通路,促進活性氧產生,導致鈣結節形成[18]。這表明,氧化應激與 AVIC 表型轉換及 AS 進展密切相關,若從抑制活性氧堆積和降低氧化應激水平的角度出發,極有可能探索出可緩解 AVIC 活化乃至 AS 狹窄進程的潛在靶點。另有研究報道,在 AS 患者切除的瓣膜中檢測到氧化低密度脂蛋白增加,而 PGC-1α 的表達降低可增加氧化低密度脂蛋白誘導的活性氧堆積及所致的單核細胞炎癥反應;反之,PGC-1α 的表達增加則可增強自噬,減輕真皮成纖維細胞向肌成纖維細胞轉變[19]。因此,我們推測 PGC-1α 可能與 AVIC 表型轉變進程相關,并選擇該靶分子進行后續實驗。
本研究發現,大鼠 AVIC 活化表型中,伴隨活化標志物骨膜蛋白、CTGF 和 α-SMA 表達明顯上調,PGC-1α 表達水平發生顯著下降。PGC-1α 參與控制多個系統(尤其是心血管系統)的脂質代謝和氧化應激途徑[20]。在心臟中,作為氧化代謝的關鍵調節因子,沉默信息調節因子 1-PGC-1α 軸可通過減少炎癥、氧化應激、降低纖維化而發揮心臟保護作用[21],若抑制 PGC-1α 活性則會加重心功能不全和心力衰竭的臨床癥狀[22],在 PGC-1α 缺乏的小鼠中,可觀察小鼠左心室功能障礙和心率異常[23];在血管中,在臨床標本及實驗動物的動脈粥樣硬化模型中均表達下調,而激活 PGC-1α 則可發揮抗動脈粥樣硬化作用[24];此外,PGC-1α 參與了多種纖維化疾病的細胞表型轉換,如在各種急、慢性腎病及老齡引起的腎纖維化動物模型中,腎組織內 PGC-1α 高表達可緩解腎纖維化,而敲除 PGC-1α 基因的動物出現嚴重的腎纖維化[25];在心肌成纖維細胞中特異性敲除 PGC-1α 可誘發心肌重塑,加重血管緊張素Ⅱ誘導的心肌纖維化,伴隨氧化應激反應和炎癥反應出現[5]。但是,目前尚無關于 PGC-1α 與 AS 的相關研究。因此我們進行了相關研究,且結果顯示,在大鼠和人 AVIC 活化表型中,PGC-1α 的表達增加均發揮了保護性作用,可以緩解 TGF-β1 誘導的 AVIC 活化程度。但是其作用機制是否如前所述與活性氧的生成減少有關及其如何調控活性氧生成均尚未可知。
PGC-1α 基因位于小鼠的 5 號染色體(人類的 4 號染色體)上,編碼含有 797 個(小鼠)或 798 個(人類)氨基酸的蛋白質。PGC-1α C 端包含絲氨酸/精氨酸和 RNA 識別基序結構域,可通過識別靶基因上特定序列,與各種轉錄因子和核受體結合來發揮其轉錄調節功能[26]。肝細胞中免疫共沉淀后轉錄組分析報道顯示,可被 PGC-1α 潛在結合而調控的 RNA 為編碼 FOXO1、PER1、TRPC5 和 CAMK1δ 等[7]。因此,我們通過體外細胞實驗進行了篩選驗證,結果發現過表達 PGC-1α 能下調 CAMK1δ 的表達水平,而沉默 CAMK1δ 的表達后 AVIC 纖維性活化程度明顯減輕,表明 PGC-1α 至少部分通過調控 CAMK1δ 的表達水平發揮 AVIC 活化抑制作用。CAMK1δ 是 CAMK1 家族的成員,CAMK1 亞型包括 CAMK1α、β、γ 和 δ。可被上游 CAMK 激酶磷酸化其蘇氨酸殘基而激活,介導細胞信號轉導[27]。有研究表明,衰老是 AS 的主要危險因素,可通過 NADPH 氧化酶 4-活性氧-CAMK 通路介導心臟重塑[28]。但目前,關于 CAMK1δ 與成纖維樣細胞表型轉變的研究較少,其細胞內的功能作用尚未完全闡明,后續仍待深入探究。
活性氧是一大類氧化劑的統稱,包括源自氧的分子,如超氧化物、過氧化氫、羥基自由基、臭氧和單線態氧等。活性氧在許多心臟病的發病機制中起著關鍵作用,包括 AS、血管鈣化、動脈粥樣硬化、心臟纖維化重塑等,并且通常與 TGF-β 信號傳導有關[29]。且如前所述,PGC-1α 具有抑制活性氧生成的作用,并且 PGC-1α 可下調 CAMK1δ 表達,而 CAMK1δ 上調與活性氧增加有關[30]。系列證據表明,在 AVIC 活化進程中,PGC-1α 通過下調 CAMK1δ 表達水平抑制活性氧生成而發揮 AVIC 活化抑制作用。因此,我們進行了相關實驗,并且驗證出 PGC-1α 在 AVIC 活化進程中至少部分通過減少活性氧生成而發揮作用。
mTOR 是一種非典型絲氨酸/蘇氨酸激酶,以 mTOR 復合體 1(mTORC1)和 mTOR 復合體 2(mTORC2)的形式存在,屬于磷脂酰肌醇激酶家族,在調控細胞凋亡、生長、增殖和自噬中起著至關重要的作用[31]。既往研究報道,在肺纖維化疾病模型中,TGF-β-PI3K-AKT-mTOR 信號通路介導脂多糖誘導的肺成纖維細胞活化[32]。mTOR 被上游調控因子調節,活性氧是其初始調節因子之一[31]。因此,后續我們驗證了 PGC-1α 的作用是否通過此通路進行。結果發現,過表達 PGC-1α 后,伴隨 AVIC 活化程度緩解、CAMK1δ 表達水平下調、活性氧活性下降的同時,p-mTOR 通路激活受到抑制,提示我們 PGC-1α 至少部分通過抑制 p-mTOR 信號通路激活發揮 AVIC 活化抑制作用。
綜上所述,本研究結果表明,過表達 PGC-1α 能通過下調 CAMK1δ 的表達、抑制活性氧活性、負調控 p-mTOR 信號通路激活,進而緩解 TGF-β1 誘導的 AVIC 活化程度,即 PGC-1α 在 AVIC 乃至 AS 進程中發揮保護性作用,有望成為 AS 疾病治療的靶點。
利益沖突:所有作者聲明不存在利益沖突。
主動脈瓣狹窄(aortic stenosis, AS)是先天性因素如瓣葉發育畸形或后天性因素如退行性變、風濕性變誘發的主動脈瓣進行性重塑出現纖維化、鈣化,而造成主動脈瓣葉增厚粘連、瓣口狹窄、左心室流出道受阻進而導致左心室不良重塑的疾病[1]。AS 是最常見的心臟瓣膜病,發病率及病死率高,但目前尚無有效的早期干預措施及治療藥物。因此,對 AS 的發病機制進行深入研究,并推動潛在藥物治療靶點的發現顯得尤為重要。AS 誘因多樣、發病機制復雜。其中,主動脈瓣的主要細胞成分主動脈瓣瓣膜間質細胞(aortic valve interstitial cells, AVIC)發生活化并向成骨分化被認為是 AS 的關鍵環節[2]。但目前 AVIC 的活化機制尚未闡明。臨床前和人體研究數據表明,各種來源的活性氧的不良積累介導了 AS 起始階段的浸潤性脂質的氧化和炎癥反應,進而誘導 AVIC 出現肌成纖維細胞樣表型活化,且氧化應激水平升高程度與 AS 的嚴重程度相關[3]。據報道,脂質氧化及其介導的細胞表型轉換可受過氧化物酶體增殖物激活受體 γ 共激活因子 1α(peroxisome proliferator-activated receptor gamma coactivator 1α, PGC-1α)的調控[4]。PGC-1α 是控制多種核編碼的線粒體基因的關鍵分子,可通過激活多種轉錄因子調控驅動線粒體呼吸鏈和脂肪酸氧化酶基因的表達,促使線粒體數目增加、線粒體呼吸能力增強[5]。既往研究表明,PGC-1α 可作為氧化應激的關鍵抑制因子降低活性氧生成、減少氧化應激及炎癥反應[6]。更重要的是,PGC-1α 可通過調控核呼吸因子而調節線粒體轉錄因子 A、B1、B2 的表達繼而調節線粒體基質蛋白的表達發揮活性氧脫毒作用。PGC-1α 包含參與 RNA 調控的蛋白質結構域如絲氨酸/精氨酸和 RNA 識別基序,可直接調控 RNA 的表達,而 AVIC 活化進程中 PGC-1α 是否通過作用于特殊 RNA 而發揮作用仍尚未明確。既往 PGC-1α 免疫共沉淀后的轉錄組測序結果顯示,其可能與鈣調蛋白依賴性蛋白激酶 1δ(calcium/calmodulin-dependent protein kinase 1δ, CAMK1δ)、生物鐘基因周期蛋白 1(period 1, PER1)、經典瞬時受體電位通道 5(transient receptor potential canonical 5, TRPC5)、叉頭框蛋白 O1(fork-head box protein O1, FOXO1)等存在相互作用[7]。目前,PGC-1α 在 AS 關鍵環節即 AVIC 表型轉變中的作用及機制尚未可知。因此,本研究旨在探討 AVIC 活化進程中 PGC-1α 的表達水平改變情況、作用特點及機制途徑。
1 材料與方法
1.1 實驗試劑
高糖基礎培養基、減血清培養基、0.25% 胰蛋白酶、Lipofectamine RNAiMAX 試劑、Ⅱ型膠原酶、胎牛血清及總 RNA 抽提試劑盒 TRIzolTM均購自美國 Thermo Fisher Scientific 公司;轉化生長因子 β1(transforming growth factor β1, TGF-β1)購自北京義翹神州科技股份有限公司;聚偏二氟乙烯膜、4’, 6-二脒基-2-苯基吲哚(4’, 6-diamidino-2-phenylindole, DAPI)染液購自美國 Merck 公司;逆轉錄酶購自日本 Toyobo 公司;實時定量聚合酶鏈反應(real-time quantitative polymerase chain reaction, RT-qPCR)熒光染料預混液購自美國 BIO-RAD 公司;Triton?X-100 和 4% 多聚甲醛固定液購自中國 Biosharp 公司;活性氧檢測試劑盒(S0033)、細胞裂解液購自上海碧云天生物技術有限公司;小干擾 RNA(small interfering RNA, siRNA)由中國吉瑪基因公司合成;辣根酶標記山羊抗兔/小鼠免疫球蛋白 G 購自中國中杉金橋生物公司。抗體信息:Alexa Fluor? 488 Conjugate 熒光二抗和兔抗磷酸化哺乳動物雷帕霉素靶蛋白(phospho-mammalian target of rapamycin, p-mTOR)抗體購自美國 Cell Signaling Technology 公司;兔抗 β-微管蛋白(β-tubulin)和兔抗 β-肌動蛋白(β-actin)抗體購自武漢愛博泰克生物科技有限公司;兔 PGC-1α 抗體購自美國 Novus 公司;兔抗骨膜蛋白抗體、兔抗Ⅰ型膠原蛋白抗體、鼠抗甘油醛-3-磷酸脫氫酶(glyceraldehyde-3-phosphate dehydrogenase, GAPDH)抗體、鼠抗 α-平滑肌肌動蛋白(α-smooth muscle actin, α-SMA)抗體和兔抗 CAMK1δ 抗體均購自英國 Abcam 公司。
1.2 細胞培養及 AVIC 活化模型構建
用以提取 AVIC 的 6 周齡、體重 200~250 g 的雄性 SD 大鼠購自成都達碩實驗動物有限公司(動物操作獲四川大學華西醫院動物倫理委員會批準,批件號:20230920002)。該研究所使用的主動脈夾層患者來源的主動脈瓣已通過四川大學華西醫院生物醫學倫理委員會的批準(批準號:201793)。本研究符合赫爾辛基宣言中所述的人體組織使用原則。AVIC 接種于含有 20% 胎牛血清、1% 青霉素-鏈霉素的高糖培養基中,5% 二氧化碳孵箱中培養,待細胞融合度達 95% 左右進行傳代,并將血清減半。
作為誘導成纖維細胞活化的經典刺激,細胞因子 TGF-β1 目前也被廣泛用以刺激 AVIC 以誘導其活化[8],本實驗采用 30 ng/mL 的 TGF-β1 重組蛋白構建大鼠 AVIC 活化模型。實驗分組(n=4):① 未加 TGF-β1 的空白對照組;② 加 TGF-β1 刺激 24 h 組;③ 加 TGF-β1 刺激 48 h 組;④ 加 TGF-β1 刺激 72 h 組。通過檢測纖維化標志物結締組織生長因子(connective tissue growth factor, CTGF)[9]、骨膜蛋白[10]或Ⅰ型膠原蛋白[11]、α-SMA[12]的表達水平變化評估模型是否構建成功。
1.3 細胞轉染及分組
1.3.1 體外 AVIC 過表達重組腺病毒轉染及抑制劑干預
為探討 PGC-1α 在 AVIC 活化進程中的作用,本研究通過腺病毒轉染的方法外源特異性過表達/使用過氧化物酶體增殖物激活受體 γ(peroxisome proliferator-activated receptor γ, PPARγ)拮抗劑 GW9662 抑制大鼠 AVIC 中 PGC-1α 的表達。
本實驗均以 12 孔板細胞培養板 1 mL 體系為例進行干預。因活化標志物均在 TGF-β1 刺激 48 h 時開始顯著上調,后續體外實驗均采用 TGF-β1 誘導 AVIC 48 h 進行模型構建。轉染 PGC-1α 過表達腺病毒(ADPGC-1α)外源性上調 PGC-1α 的表達,預實驗結果顯示,與空白對照 ADC 組相比,以最佳感染復數為 300 時攜帶的 PGC-1α 過表達序列的腺病毒(ADPGC-1α 組)轉染能使 AVIC 中 PGC-1α 的蛋白和基因表達水平均顯著增加,并以該方案下的 PGC-1α 過表達效率用于后續實驗。大鼠及人源 AVIC 轉染過表達腺病毒的實驗分組(n=3):① 空白對照 ADC 組;② ADC+TGF-β1 組;③ ADPGC-1α 組;④ ADPGC-1α+TGF-β1 組。隨后使用 PPARγ 拮抗劑 GW9662 抑制大鼠 AVIC 中 PGC-1α 的生物活性[13],實驗分 4 組(n=3):① 空白對照二甲基亞砜(dimethyl sulfoxide, DMSO)組;② DMSO+TGF-β1 組;③ GW9662 組;④ GW9662+TGF-β1 組。隨后以 RT-qPCR、蛋白質印跡法、細胞免疫熒光法觀察 AVIC 活化程度以及活性氧水平的改變情況。
1.3.2 體外 AVIC 小干擾 RNA 轉染
為進一步探索 PGC-1α 調節 AVIC 活化的下游分子和下游信號通路,結合文獻報道,以 siRNA 特異性沉默大鼠 AVIC 中 CAMK1δ 的表達,分為 4 組(n=3):① 空白對照 SiNC 組;② SiNC+TGF-β1 組;③ SiCAMK1δ 組;④ SiCAMK1δ+TGF-β1 組。隨后以 RT-qPCR、蛋白質印跡法觀察 AVIC 活化程度的改變情況。
1.4 流式細胞術
本研究發現 PGC-1α 可通過下調 CAMK1δ 發揮作用,而 CAMK1δ 的下調與活性氧生成減少密切相關,那么在 AVIC 活化進程中,PGC-1α 是否可通過抑制活性氧生成而發揮作用尚未可知。而活性氧與多種細胞信號通路的激活密切相關,因此,本研究篩選了 PGC-1α 下游可能作用的信號通路激活情況,如活性氧可作用于的哺乳動物雷帕霉素靶蛋白(mechanistic target of rapamycin, mTOR)及可作用于活性氧生成的低氧誘導因子 1α(hypoxia-inducible factor-1α, HIF-1α)等。隨后以流式細胞實驗觀察大鼠 AVIC 中活性氧水平的改變情況。
通過 BD FACSCanto?流式分析儀(美國 BD 公司)檢測熒光標記的細胞內活性氧及 PGC-1α 分子。將大鼠 AVIC 傳代細胞鋪于 12 孔細胞培養板內待細胞融合度達 70%,根據實驗需求處理細胞。待收樣時棄去原培養基,分別加入稀釋的熒光探針加載 2’,7’-二氯熒光黃雙乙酸鹽試劑盒(1∶
1.5 細胞免疫熒光染色實驗
根據實驗設計,待達到細胞實驗時間節點,棄去原培養基,用磷酸鹽緩沖液(phosphate buffer saline, PBS)漂洗 3 次,室溫下加入 4% 多聚甲醛固定液固定 15~20 min 后再用 PBS 漂洗 3 次,用膠頭滴管吸凈 PBS 液,加入含有 0.5% 細胞通透劑 Triton?X-100 和 1% 牛血清蛋白的混合液,室溫封閉穿孔 60 min 后用 PBS 漂洗 3 次,3 min/次。隨后加入稀釋后的 α-SMA 一抗(1∶200),4℃孵育過夜處理。第 2 天回收一抗后用 PBS 漂洗 3 次,3 min/次。加入免疫熒光二抗后室溫下避光孵育 1 h 后用 PBS 漂洗 3 次,3 min/次。滴加 DAPI(用 PBS 稀釋為 1∶
1.6 RT-qPCR 檢測基因的表達變化
通過 RT-qPCR 對基因轉錄水平進行檢測。將 TRIzol 裂解液加入 12 孔板細胞培養板中裂解細胞,通過氯仿法提取總 RNA。通過與逆轉錄酶孵育,根據表1 配制逆轉錄反應體系,本研究中使用的引物序列見表2,將總 RNA 逆轉錄為單鏈互補 DNA。根據制造商的說明,使用試劑盒進行 RT-qPCR 檢測。在 CFX Connect 機器(美國 BIO-RAD 公司)上檢測到信號。GAPDH 或 β-actin 作為內參基因,即相對基因表達歸一化為 GAPDH 表達,進行定量分析。
1.7 蛋白質印跡法檢測蛋白表達
根據實驗設計待細胞實驗完成后,將培養的細胞直接在放射免疫沉淀試驗緩沖液中裂解,冰上裂解 15 min,4℃,
1.8 統計學方法
使用 GraphPad Prism 8.0 軟件進行統計分析,實驗結果以均數±標準差表示。兩組間比較使用兩獨立樣本 t 檢驗,多組間比較使用單因素方差分析,組間兩兩比較使用SNK-q檢驗。雙側檢驗水準 α=0.05。
2 結果
2.1 PGC-1α 表達水平在活化的大鼠 AVIC 中下降
2.1.1 TGF-β1 誘導的大鼠 AVIC 活化模型構建成功
實驗結果表明,與空白對照組相比,活化標志物骨膜蛋白的 mRNA 表達水平在 TGF-β1 刺激大鼠 AVIC 24、48、72 h 后呈梯度增加現象,CTGF 及Ⅰ型膠原蛋白的 mRNA 表達水平亦在 TGF-β1 作用 48、72 h 后表達增加(圖1a~1c),表明 TGF-β1 誘導的大鼠 AVIC 活化模型構建成功。
圖1
PGC-1α 在活化的 AVIC 中表達降低
a~d. 大鼠 AVIC 在 TGF-β1 刺激 24、48、72 h 后,各纖維化標志物及 PGC-1α 的 mRNA 表達水平(
2.1.2 PGC-1α 在 TGF-β1 誘導活化的大鼠 AVIC 中表達下調
與空白對照組(1.00±0.18)相比,TGF-β1 刺激大鼠 AVIC 24、48、72 h 后,PGC-1α 的 mRNA 表達水平均出現明顯下調(24 h:0.31±0.10;48 h:0.32±0.06;72 h:0.20±0.07),且差異有統計學意義(P<0.05,圖1d)。流式細胞術結果顯示,TGF-β1 刺激 AVIC 48 h 后,PGC-1α 的蛋白表達水平亦顯著下調(圖1e、1f)。
2.2 PGC-1α 可緩解大鼠 AVIC 活化程度
2.2.1 過表達 PGC-1α,AVIC 活化程度減輕
與對照組(ADC+TGF-β1 組)相比,過表達 PGC-1α 后(ADPGC-1α+TGF-β1 組),伴隨 PGC-1α 的表達明顯上調,TGF-β1 誘導的纖維化標志物骨膜蛋白、α-SMA 的基因和蛋白表達均明顯降低(骨膜蛋白:3.17±0.64 vs. 1.45±0.54,P<0.05;α-SMA:0.77±0.11 vs. 0.28±0.06,P<0.05),見圖2a~2h。流式細胞術和免疫熒光結果均驗證了高表達 PGC-1α 后,TGF-β1 誘導的 α-SMA 表達水平出現明顯降低(圖2i~2k)。
圖2
PGC-1α 可緩解大鼠 AVIC 活化程度
a~d. 轉染 ADPGC-1α 前后各組大鼠 AVIC 的 PGC-1α 及骨膜蛋白、α-SMA、Ⅰ型膠原蛋白的 mRNA 表達水平(
2.2.2 抑制 PGC-1α 活性,AVIC 活化程度增加
進一步反向驗證 PGC-1α 在 AVIC 活化進程中的作用發現,GW9662 抑制 PGC-1α 活性可進一步上調 TGF-β1 所誘導的骨膜蛋白表達水平,差異有統計學意義(2.20±0.68 vs. 7.99±2.50,P<0.05),同時 α-SMA 表達也呈現出上調趨勢(1.25±0.18 vs. 1.56±0.07,P=0.067)。見圖2l~2n。
2.3 PGC-1α 通過下調 CAMK1δ 的表達發揮作用
過表達 PGC-1α 表達水平后進行下游篩選,鑒定到 PGC-1α 可能通過下調 CAMK1δ 的表達發揮作用(0.97±0.04 vs. 0.74±0.11,P<0.05)(圖3a~3g)。隨后在 TGF-β1 誘導下,使用 Si CAMK1δ 特異性沉默其表達水平后觀察 AVIC 活化程度改變情況,結果顯示,沉默 CAMK1δ 表達下調了活化標志物骨膜蛋白的表達(1.76±0.11 vs. 0.99±0.20,P<0.05),表明 AVIC 活化程度緩解(圖3h~3j)。
圖3
PGC-1α 可通過下調 CAMK1δ 表達水平發揮作用
a~g. 轉染 ADPGC-1α 前后各組大鼠 AVIC 的骨膜蛋白及
2.4 PGC-1α 抑制活性氧生成而發揮作用
流式細胞學結果顯示,TGF-β1 作用下,與對照組(ADC+TGF-β1 組)相比,ADPGC-1α+TGF-β1 組活性氧活性降低且差異有統計學意義(778.3±139.4 vs. 159.3±43.2,P<0.05)(圖4a、4b)。同時,功能抑制實驗結果顯示,TGF-β1 刺激下,與對照組(DMSO+TGF-β1 組)相比,GW9662 的使用(GW9662+TGF-β1 組)進一步增加了 TGF-β1 誘導的 AVIC 活化進程中的活性氧水平(圖4c、4d)。
圖4
PGC-1α 抑制活性氧生成而發揮作用
a、b. 存在或不存在 TGF-β1 刺激的情況下,AVIC 轉染 ADPGC-1α 后活性氧的流式細胞術結果(
2.5 PGC-1α 負調控 p-mTOR 通路激活而發揮作用
蛋白質印跡法結果顯示,TGF-β1 刺激下,與對照組(ADC+TGF-β1)相比,ADPGC-1α+TGF-β1 組 p-mTOR 的表達明顯下調(圖5a);相反,使用 GW9662 抑制 PGC-1α 活性后,p-mTOR 通路分子表達上調(圖5b);HIF-1α 則無明顯變化。
圖5
PGC-1α 可負調控 p-mTOR 通路激活而發揮作用
a. 存在或不存在 TGF-β1 刺激的情況下,AVIC 轉染 ADPGC-1α 后通路蛋白分子 p-mTOR、HIF-1α 的蛋白水平(
鑒于 PGC-1α 可下調 CAMK1δ 表達,而文獻報道 CAMK1δ 表達的下降與活性氧生成減少相關,隨后,我們觀察了特異性沉默 CAMK1δ 后 mTOR 通路的激活情況,結果顯示,下調 CAMK1δ 表達水平后 mTOR 通路激活程度減輕(圖5c)。
2.6 過表達 PGC-1α 緩解 TGF-β1 誘導的人源的 AVIC 活化程度
基于上述結果,通過人來源的 AVIC 進行驗證 PGC-1α 發揮作用的物種保守性。蛋白質印跡實驗結果顯示,與對照組(ADC+TGF-β1 組)相比,ADPGC-1α+TGF-β1 組活化標志物骨膜蛋白、CTGF 蛋白水平表達均明顯降低(骨膜蛋白:2.73±0.53 vs. 1.63±0.14,P<0.05;CTGF:1.27±0.04 vs. 0.48±0.09,P<0.05),差異有統計學意義(圖6)。
圖6
過表達 PGC-1α 可緩解 TGF-β1 誘導的人源的 AVIC 活化程度
a~d. 存在或不存在 TGF-β1 刺激的情況下,人源 AVIC 轉染 ADPGC-1α 后骨膜蛋白、α-SMA 的蛋白表達水平(
3 討論
AS 主要病理表現為主動脈瓣葉體部纖維化鈣化增厚、主動脈瓣環狹窄、左心室的機械應力增加引起嚴重并發癥,目前尚無有效緩解藥物[14]。經導管主動脈瓣置換術是重度 AS 的成熟治療手段,但術后傳導阻滯、急性腎損傷、卒中等并發癥及瓣膜衰敗等后續不良后果的出現,使 AS 發病機制的探索及藥物治療靶點的發現變得尤為重要。
AS 誘因多樣、發病機制復雜,分子和細胞過程未被清楚表征。現有研究認為,瓣膜內皮功能受損、脂質浸潤沉積、炎癥細胞滲出、免疫炎癥反應以及在此基礎上發生的瓣膜纖維化和/或鈣化是其主要機制[15]。在此過程中 AVIC 的表型轉變包括纖維性活化及向成骨分化(即鈣化)是 AS 的關鍵環節。盡管所有終末期 AS 瓣膜都表現為鈣化,但纖維化在 AS 中始終起著重要作用[16]。因此,本研究選擇 AVIC 作為靶細胞,研究 AS 早期細胞表型轉換(即 AVIC 活化)的生物學機制,以期為輕度 AS 患者進展為重度 AS 患者的中間過程提供潛在阻遏靶點。
如前所述,一項使用硫代巴比妥酸評估全身脂質過氧化和 2, 4-二硝基苯肼評估血漿蛋白氧化修飾的隊列研究表明,氧化應激水平升高與 AS 的嚴重程度相關[17]。在分離培養的豬 AVIC 中,TGF-β1 刺激可通過激活促分裂素原活化蛋白激酶信號通路,促進活性氧產生,導致鈣結節形成[18]。這表明,氧化應激與 AVIC 表型轉換及 AS 進展密切相關,若從抑制活性氧堆積和降低氧化應激水平的角度出發,極有可能探索出可緩解 AVIC 活化乃至 AS 狹窄進程的潛在靶點。另有研究報道,在 AS 患者切除的瓣膜中檢測到氧化低密度脂蛋白增加,而 PGC-1α 的表達降低可增加氧化低密度脂蛋白誘導的活性氧堆積及所致的單核細胞炎癥反應;反之,PGC-1α 的表達增加則可增強自噬,減輕真皮成纖維細胞向肌成纖維細胞轉變[19]。因此,我們推測 PGC-1α 可能與 AVIC 表型轉變進程相關,并選擇該靶分子進行后續實驗。
本研究發現,大鼠 AVIC 活化表型中,伴隨活化標志物骨膜蛋白、CTGF 和 α-SMA 表達明顯上調,PGC-1α 表達水平發生顯著下降。PGC-1α 參與控制多個系統(尤其是心血管系統)的脂質代謝和氧化應激途徑[20]。在心臟中,作為氧化代謝的關鍵調節因子,沉默信息調節因子 1-PGC-1α 軸可通過減少炎癥、氧化應激、降低纖維化而發揮心臟保護作用[21],若抑制 PGC-1α 活性則會加重心功能不全和心力衰竭的臨床癥狀[22],在 PGC-1α 缺乏的小鼠中,可觀察小鼠左心室功能障礙和心率異常[23];在血管中,在臨床標本及實驗動物的動脈粥樣硬化模型中均表達下調,而激活 PGC-1α 則可發揮抗動脈粥樣硬化作用[24];此外,PGC-1α 參與了多種纖維化疾病的細胞表型轉換,如在各種急、慢性腎病及老齡引起的腎纖維化動物模型中,腎組織內 PGC-1α 高表達可緩解腎纖維化,而敲除 PGC-1α 基因的動物出現嚴重的腎纖維化[25];在心肌成纖維細胞中特異性敲除 PGC-1α 可誘發心肌重塑,加重血管緊張素Ⅱ誘導的心肌纖維化,伴隨氧化應激反應和炎癥反應出現[5]。但是,目前尚無關于 PGC-1α 與 AS 的相關研究。因此我們進行了相關研究,且結果顯示,在大鼠和人 AVIC 活化表型中,PGC-1α 的表達增加均發揮了保護性作用,可以緩解 TGF-β1 誘導的 AVIC 活化程度。但是其作用機制是否如前所述與活性氧的生成減少有關及其如何調控活性氧生成均尚未可知。
PGC-1α 基因位于小鼠的 5 號染色體(人類的 4 號染色體)上,編碼含有 797 個(小鼠)或 798 個(人類)氨基酸的蛋白質。PGC-1α C 端包含絲氨酸/精氨酸和 RNA 識別基序結構域,可通過識別靶基因上特定序列,與各種轉錄因子和核受體結合來發揮其轉錄調節功能[26]。肝細胞中免疫共沉淀后轉錄組分析報道顯示,可被 PGC-1α 潛在結合而調控的 RNA 為編碼 FOXO1、PER1、TRPC5 和 CAMK1δ 等[7]。因此,我們通過體外細胞實驗進行了篩選驗證,結果發現過表達 PGC-1α 能下調 CAMK1δ 的表達水平,而沉默 CAMK1δ 的表達后 AVIC 纖維性活化程度明顯減輕,表明 PGC-1α 至少部分通過調控 CAMK1δ 的表達水平發揮 AVIC 活化抑制作用。CAMK1δ 是 CAMK1 家族的成員,CAMK1 亞型包括 CAMK1α、β、γ 和 δ。可被上游 CAMK 激酶磷酸化其蘇氨酸殘基而激活,介導細胞信號轉導[27]。有研究表明,衰老是 AS 的主要危險因素,可通過 NADPH 氧化酶 4-活性氧-CAMK 通路介導心臟重塑[28]。但目前,關于 CAMK1δ 與成纖維樣細胞表型轉變的研究較少,其細胞內的功能作用尚未完全闡明,后續仍待深入探究。
活性氧是一大類氧化劑的統稱,包括源自氧的分子,如超氧化物、過氧化氫、羥基自由基、臭氧和單線態氧等。活性氧在許多心臟病的發病機制中起著關鍵作用,包括 AS、血管鈣化、動脈粥樣硬化、心臟纖維化重塑等,并且通常與 TGF-β 信號傳導有關[29]。且如前所述,PGC-1α 具有抑制活性氧生成的作用,并且 PGC-1α 可下調 CAMK1δ 表達,而 CAMK1δ 上調與活性氧增加有關[30]。系列證據表明,在 AVIC 活化進程中,PGC-1α 通過下調 CAMK1δ 表達水平抑制活性氧生成而發揮 AVIC 活化抑制作用。因此,我們進行了相關實驗,并且驗證出 PGC-1α 在 AVIC 活化進程中至少部分通過減少活性氧生成而發揮作用。
mTOR 是一種非典型絲氨酸/蘇氨酸激酶,以 mTOR 復合體 1(mTORC1)和 mTOR 復合體 2(mTORC2)的形式存在,屬于磷脂酰肌醇激酶家族,在調控細胞凋亡、生長、增殖和自噬中起著至關重要的作用[31]。既往研究報道,在肺纖維化疾病模型中,TGF-β-PI3K-AKT-mTOR 信號通路介導脂多糖誘導的肺成纖維細胞活化[32]。mTOR 被上游調控因子調節,活性氧是其初始調節因子之一[31]。因此,后續我們驗證了 PGC-1α 的作用是否通過此通路進行。結果發現,過表達 PGC-1α 后,伴隨 AVIC 活化程度緩解、CAMK1δ 表達水平下調、活性氧活性下降的同時,p-mTOR 通路激活受到抑制,提示我們 PGC-1α 至少部分通過抑制 p-mTOR 信號通路激活發揮 AVIC 活化抑制作用。
綜上所述,本研究結果表明,過表達 PGC-1α 能通過下調 CAMK1δ 的表達、抑制活性氧活性、負調控 p-mTOR 信號通路激活,進而緩解 TGF-β1 誘導的 AVIC 活化程度,即 PGC-1α 在 AVIC 乃至 AS 進程中發揮保護性作用,有望成為 AS 疾病治療的靶點。
利益沖突:所有作者聲明不存在利益沖突。