

引用本文: 葛玲, 高群婷, 楊燦, 李學斌, 劉瑞寒, 李秋波, 孔慶霞. RHOBTB2 基因變異所致發育性癲癇性腦病-64型一例并文獻復習. 癲癇雜志, 2024, 10(6): 542-546. doi: 10.7507/2096-0247.202409003 復制
RHOBTB2基因與發育性癲癇性腦病-64型(developmental and epileptic encephalopathy-64,DEE-64)相關,DEE-64以癲癇發作、嚴重智力障礙、運動功能受損、出生后小頭畸形為主要表現。發育性和癲癇性腦病(developmental and epileptic encephalopathy,DEE)是一組臨床和遺傳異質的年齡依賴性神經系統疾病[1],其特征是在嬰兒期或兒童早期出現難治性癲癇發作,伴有精神運動發育遲緩或倒退。截至2023年3月,在線人類孟德爾遺傳(online mendelian inheritance in man,OMIM)數據庫收錄DEE致病基因已達110個,其中常染色體基因占比超過90%[2]。2016年首次報道RHOBTB2基因變異引起的一例以重度神經發育障礙為特征的DEE,2018年在RHOBTB2的broad-complex,tramtrack,and bric-à-brac(BTB)結構域區域中聚集的新生雜合錯義突變所致DEE[3]。本文報道一例RHOBTB2變異所致DEE病例,該患兒嬰兒期有多次抽搐病史,表現為全面性或局灶性癲癇發作及嬰兒期喂養困難,服用抗癲癇發作藥物治療效果良好,發作間期腦電圖(electroencephalogram,EEG)顯示多灶性放電,頭顱磁共振成像(magnetic resonance imaging,MRI)提示雙側額顳部腦外間隙增寬。該研究獲得濟寧醫學院附屬醫院醫學倫理委員會審核批準及患兒監護人知情同意。
病例資料 患兒 女,3月齡余。于2023年7月5日因“間斷抽搐8天”入院。患兒入院前8天出現抽搐,表現為意識喪失、呼之不應、雙目凝視、面色發紺、四肢抽搐、口吐泡沫,歷時1~6 min,1次/d~1次/h不等,抽搐間期精神可,抽搐緩解后完善腦電圖未恢復至基線狀態,無其他伴隨癥狀,病后予以適當補液治療。入院前再次出現頻繁抽搐,每次持續約1 h緩解,緩解后精神略差,進食不佳。初步診斷為“癲癇”。查體未見明顯發育異常,完善格塞爾發展量表(GESELL)結果提示發育商87(發育正常兒童發育商>70),未見明顯發育遲緩。初予以左乙拉西坦口服液及苯巴比妥片治療3周仍有抽搐,加用托吡酯后癲癇控制良好,后于2023年10月減停左乙拉西坦口服液,未在有明顯發作。5月齡余(2023年9月17日)隨訪完善GESELL表結果提示發育商43.6(<70,達發育遲緩診斷標準),存在發育遲緩,各部分能區均不同程度落后,豎頭欠佳,僅可咿呀發聲,無明顯小頭畸形表現,不會叫爸爸媽媽,復查腦電圖大致正常。1歲3月齡(2024年6月29日)末次隨訪,頭圍42.0 cm,體重8.0 kg,存在小頭畸形、全面發育落后表現,復查腦電圖大致正常。出生后因“黃疸”住院治療,期間輸注血液制品(具體不詳)。家族史未見異常。其他無特殊。
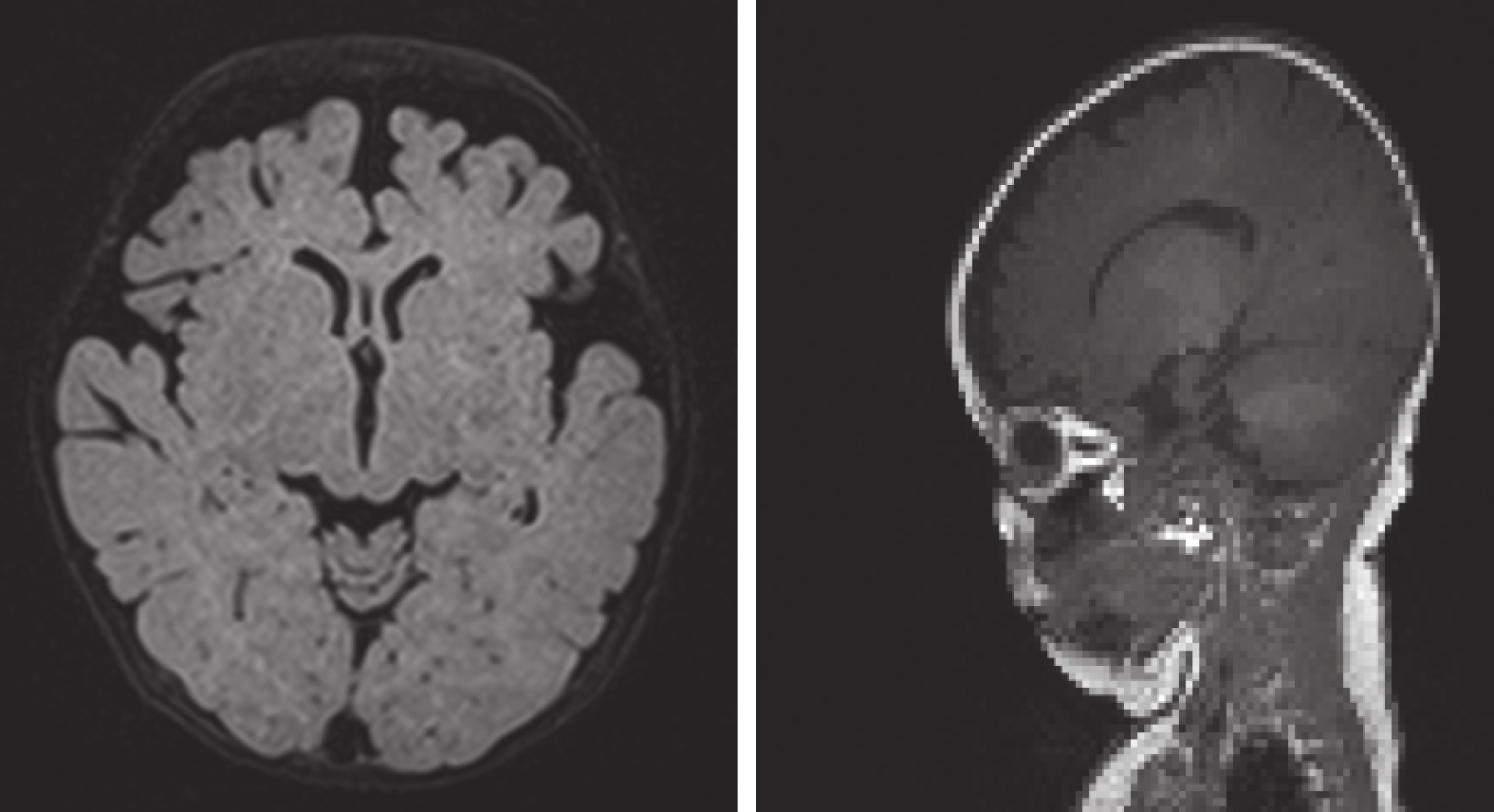
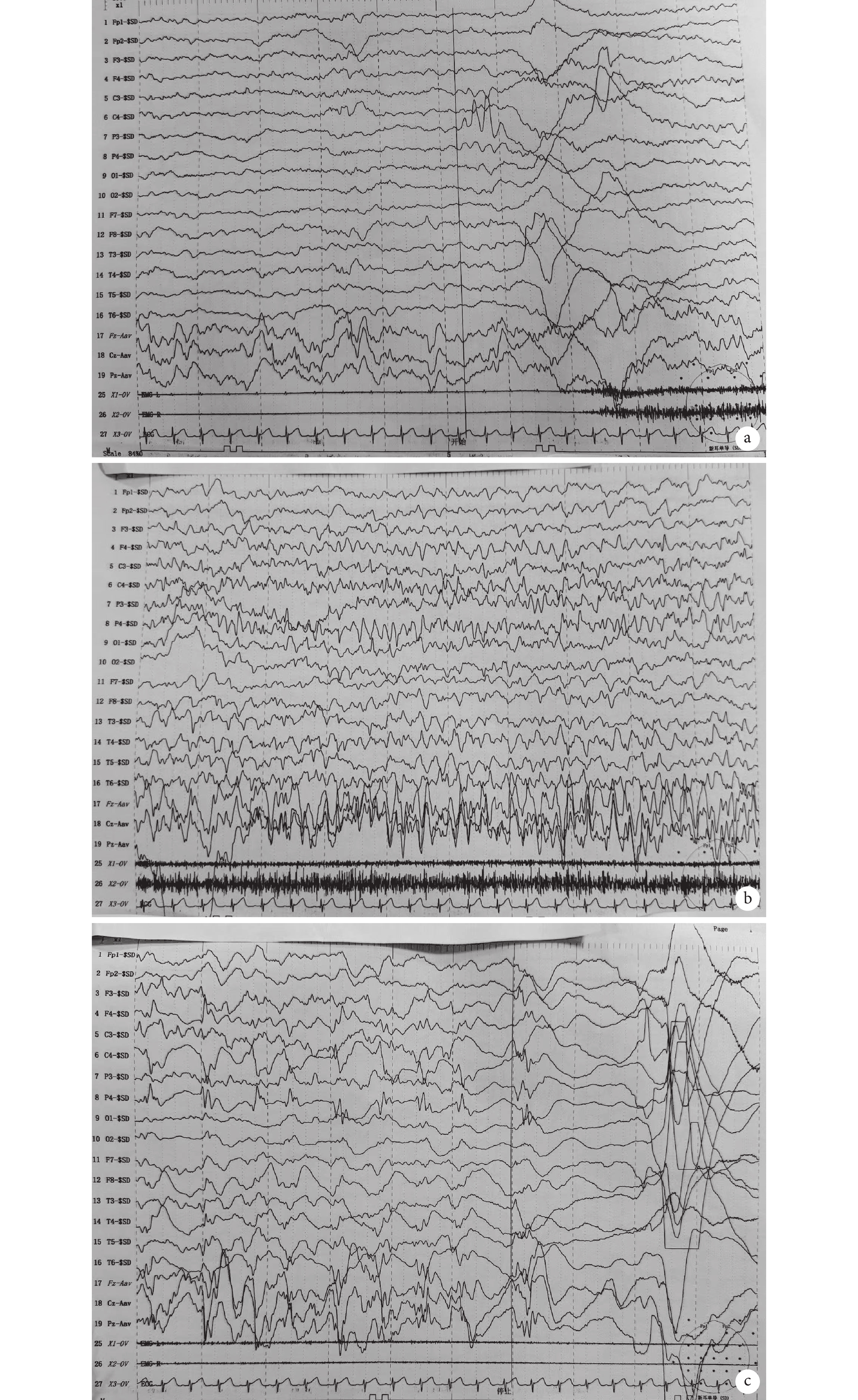
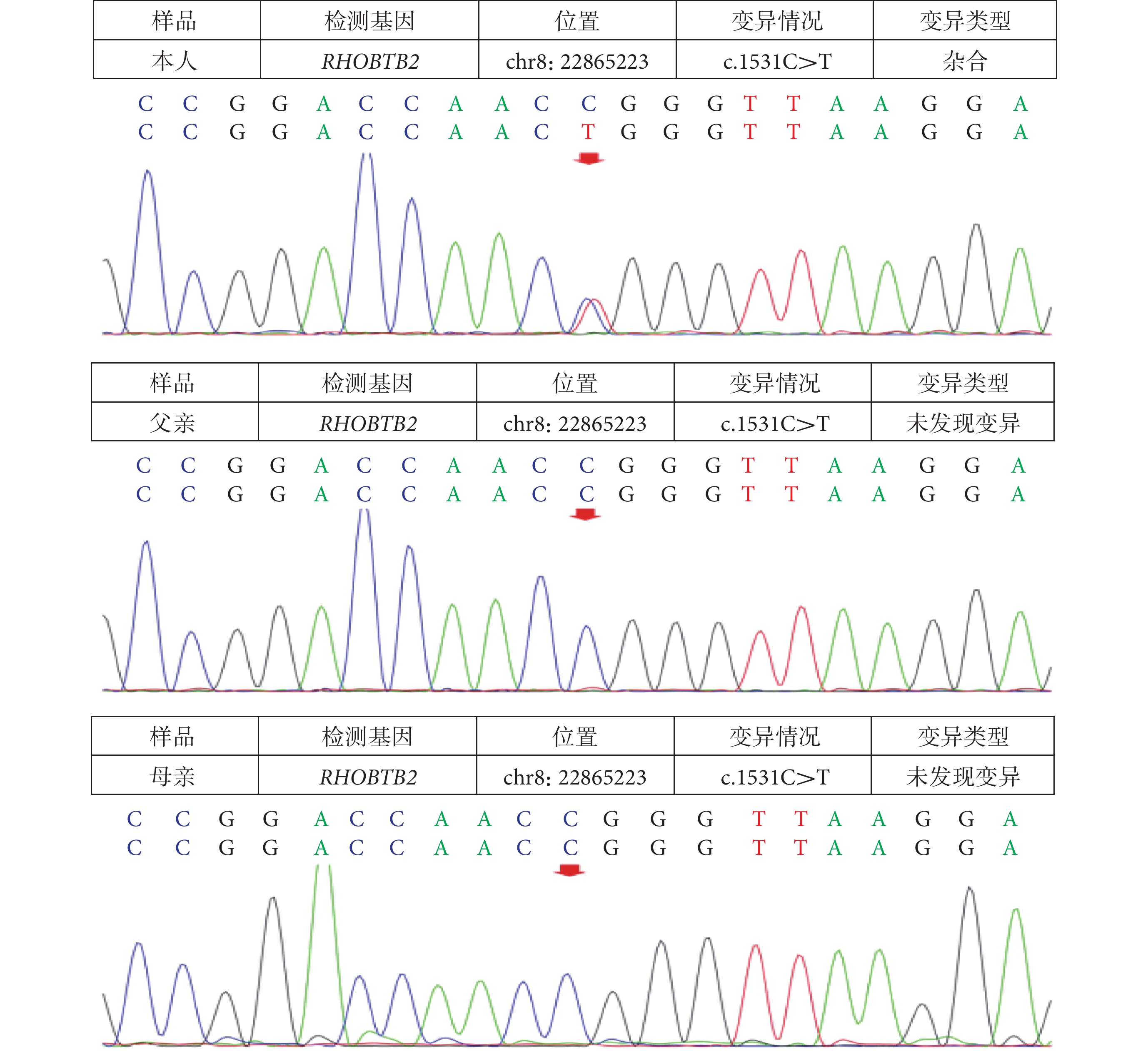
輔助檢查 血生化:電解質、肝功能、腎功能、血氨、乳酸正常; 血液及尿液代謝病篩查未見異常;酮體9.5 ng/dL。2023年7月6日頭顱MRI平掃+彌散加權成像+癲癇特殊序列示:雙側海馬對稱,未見明顯異常信號影。雙側大腦半球結構對稱,腦灰白質對比正常,腦實質內未見異常信號灶,幕下小腦半球及腦干信號未見明顯異常。雙側額顳部腦外間隙增寬,余各腦室、腦池、腦裂及腦溝未見異常,中線結構居中。結論:① 腦內結構未見明確異常信號;② 雙側額顳部腦外間隙增寬(圖1)。2023年6月29日腦電圖示:嬰兒正常范圍睡眠腦電圖、腦地形圖。2023年7月6日床旁視頻腦電圖示:異常嬰兒視頻腦電圖睡眠期雙側額區、中央區不典型尖波散發記錄到6次起源于雙側前頭部局灶性發作并泛化(圖2)。后多次復查均提示異常嬰兒床旁視頻腦電圖,多灶性放電(較前好轉)。2023年9月3日 6 h視頻腦電圖示:大致正常嬰兒視頻腦電圖。全外顯子基因測序示:患兒RHOBTB2基因:c.1531C>T即該基因
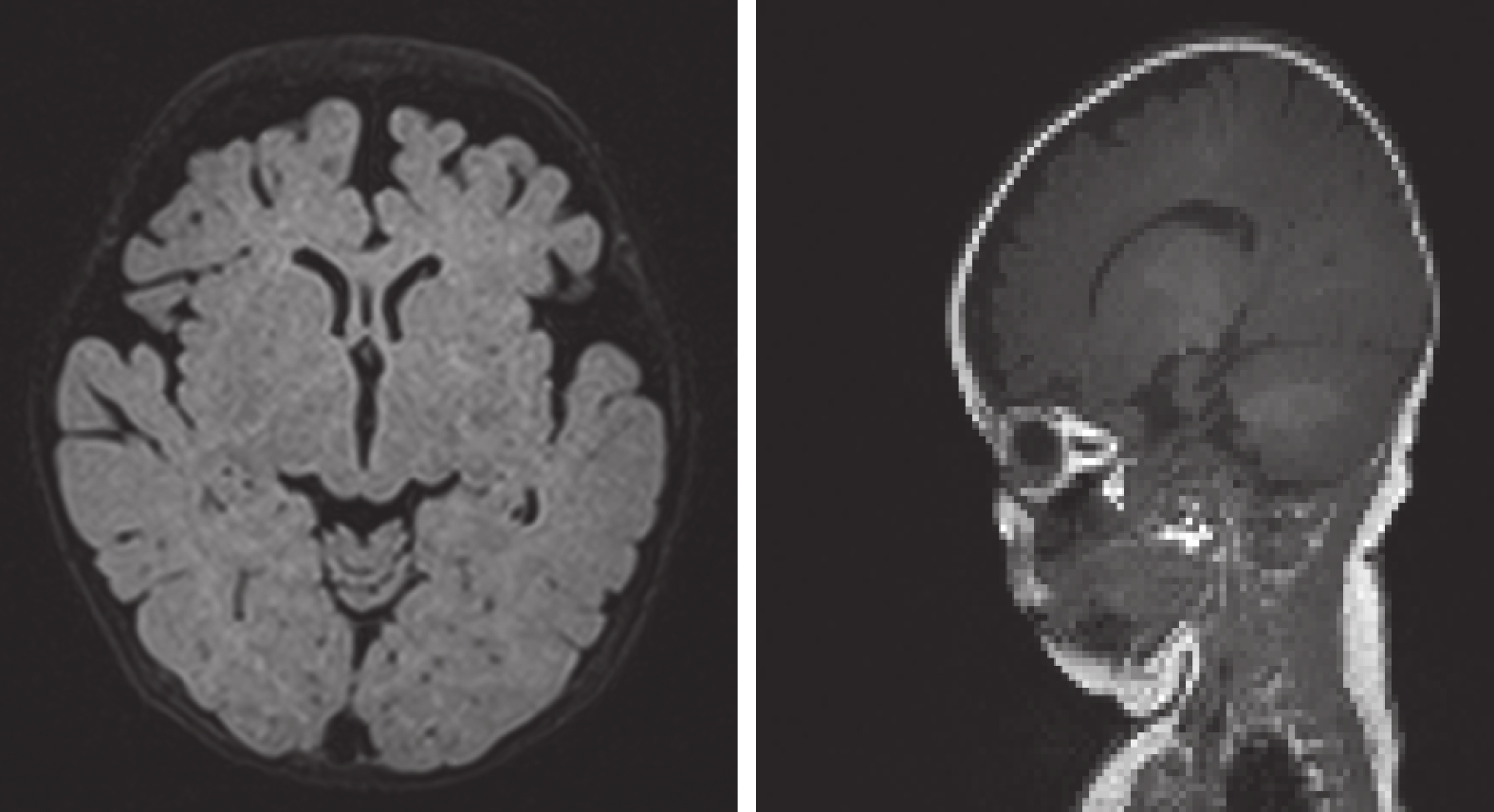 圖1
患兒頭顱磁共振成像平掃+彌散加權成像+癲癇特殊序列
圖1
患兒頭顱磁共振成像平掃+彌散加權成像+癲癇特殊序列
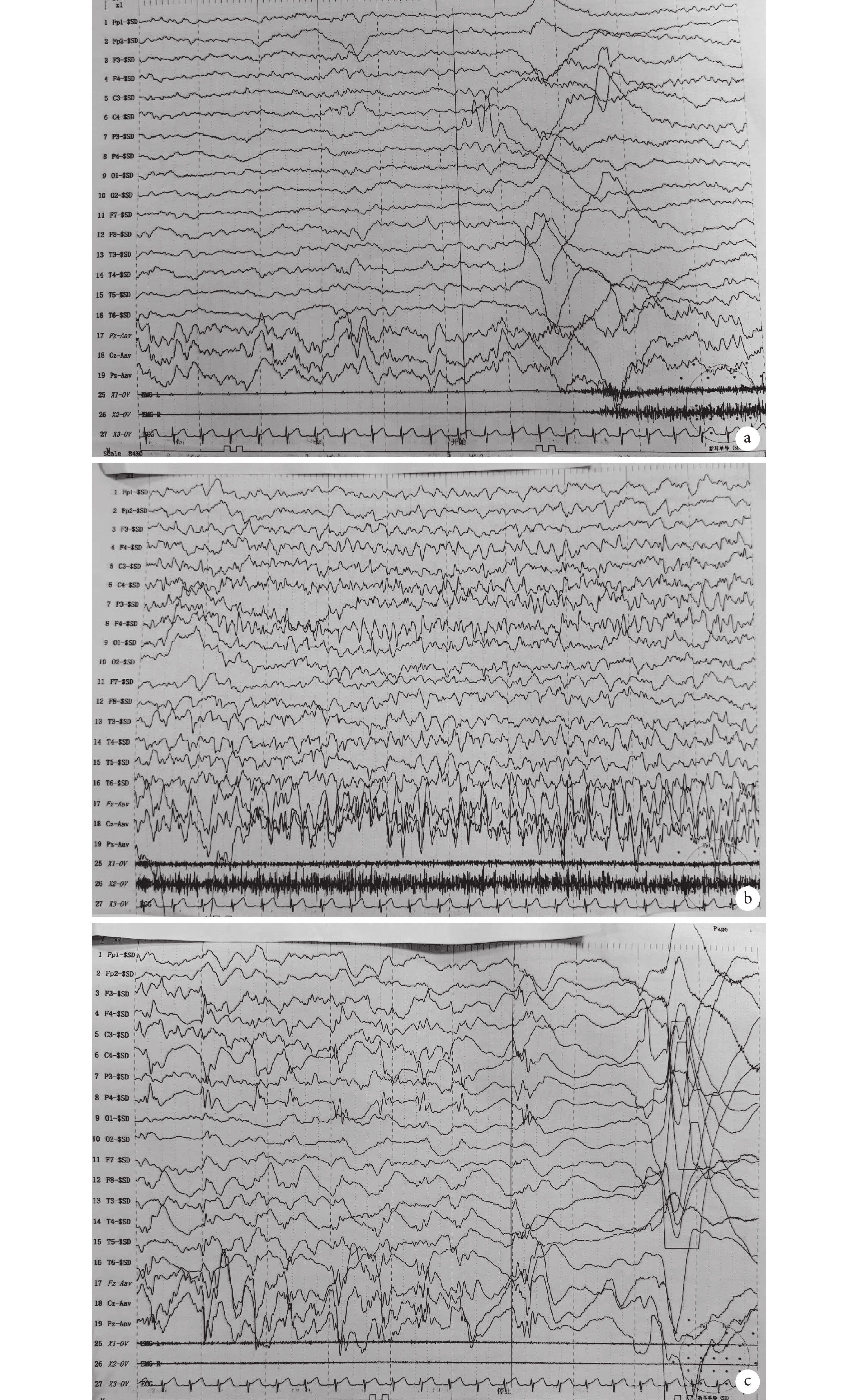 圖2
患兒腦電圖
圖2
患兒腦電圖
a. 發作初期;b. 發作期;c. 發作終止
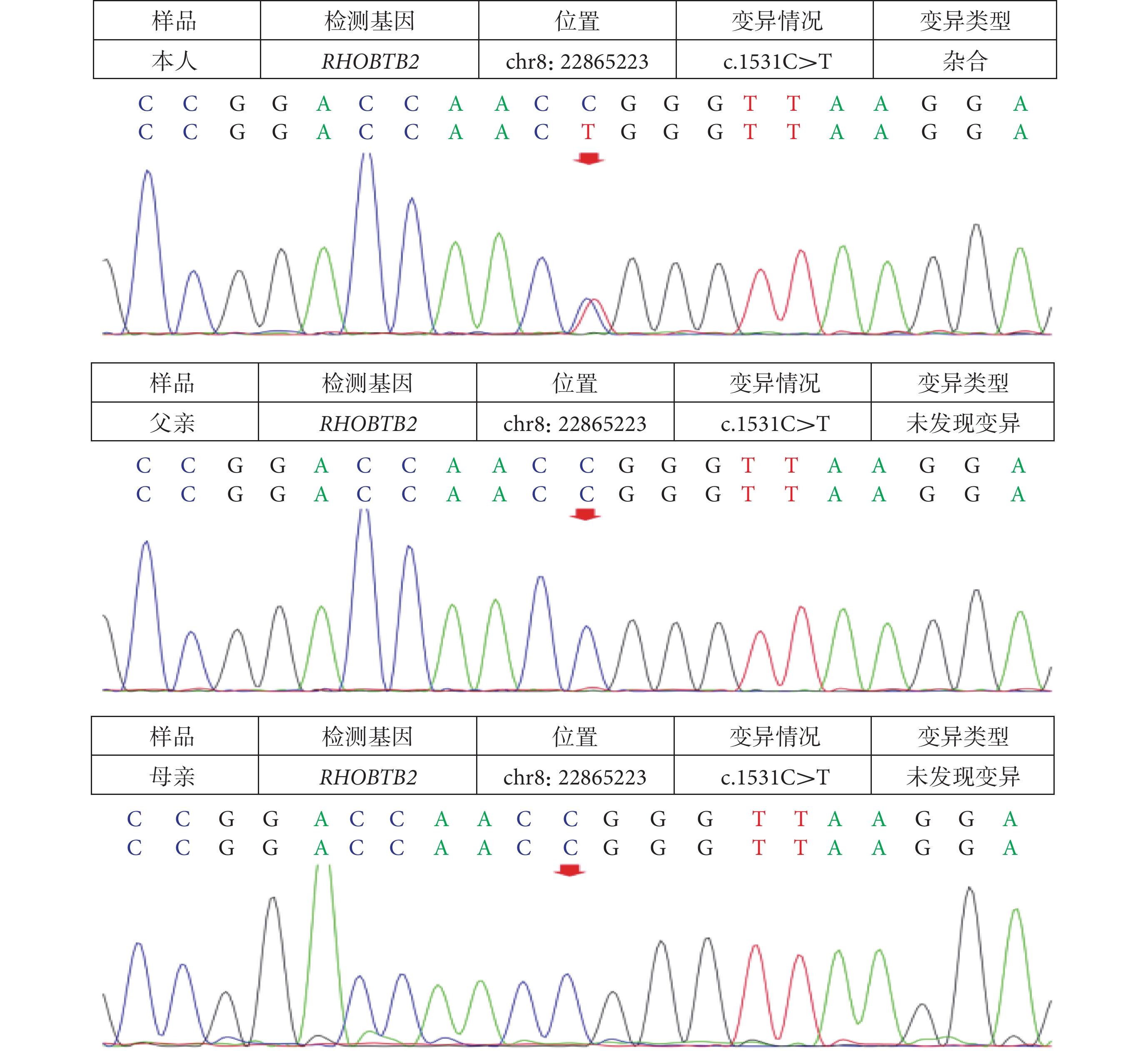 圖3
全外顯子基因測序
圖3
全外顯子基因測序
討論 本例患兒以早發性癲癇發作、癲癇持續狀態、發育遲緩為主要表現,輔助檢查提示無明顯感染指征,生化、尿液代謝指標不支持代謝性疾病,顱腦MRI不支持結構性病變所致,結合基因檢測結果符合RHOBTB2基因相關性DEE。該基因是含 Rho 相關BTB 結構域的蛋白構成非典型RhoGTP酶的一個亞家族,在哺乳動物中以RHOBTB1、RHOBTB2和RHOBTB3為代表[5]。其中RHOBTB2在神經系統中表達最豐富。RHOBTB2基因隸屬于致病基因中的酶/酶調節劑中的信號轉導酶類別,位于人類染色體8p21.3,該基因編碼的蛋白質是一種小的非典型Rho GTP酶,含有一個GTPase 結構域和2個BTB結構域。它通過BTB結構域與Culli-3依賴性泛素連接酶復合物相互作用,介導其自身的泛素化,并招募其他底物到復合物中,是細胞信號傳遞相關基因,參與各種細胞活動,如轉錄調節和蛋白質降解等。研究表明,在RHOBTB2的BTB結構域編碼區聚集的新發錯義變異的個體表現為相當均勻、嚴重的DEE,包括早發性癲癇、智力障礙、小頭畸形、陣發性運動障礙和MRI異常,與本例患兒所出現的早發性癲癇、智力障礙、小頭畸形、陣發性運動障礙高度一致[6,7]。同時Straub等[8]研究發現,Cullin-3結合位點位于第二個BTB結構域但與該p.Arg511Trp變體的位點相距甚遠,該基因位點突變不會破壞RHOBTB2的Cullin-3結合位點,且在既往報道的28例表型包括癲癇、中度至重度智力障礙的RHOBTB2新發錯義變異患者中,所有突變位點都聚集在或接近第一或第二BTB結構域,這更表明可能不是由于泛素連接酶支架蛋白cullin-3的直接相互作用介導的,而是由BTB結構域穩定性受損或二聚體形成介導的[7,9]。此外,敲低RHOBTB2基因表達量會導致樹突發育大小、長度顯著減少,導致突觸興奮與抑制失衡,RHOBTB2的過表達也會導致癲癇發作的易感性增加、運動障礙及增加促凋亡蛋白的水平,因此,DEE的病理機制也可能是由于突變體RHOBTB2水平增高增加了細胞凋亡所導致[6]。據統計,目前DEE的治療主要以對癥治療為主,多數患者口服2種或2種以上藥物治療癲癇和運動障礙,其中卡馬西平、左乙拉西坦等對運動障礙可能更有效[10],托吡酯和氟桂利嗪聯合用藥可能會降低運動障礙的發生頻率[11]。本例患兒的治療主要是口服苯巴比妥及托吡酯控制癲癇發作,目前病情控制良好,與既往文獻報道一致。
文獻復習顯示,截止2023年12月,以“(RHOBTB2)AND(Developmental epileptic encephalopathy)”為檢索詞條檢索Pubmed數據庫,共有6篇相關的英文文獻[7,8,11-14],以“RHOBTB2與癲癇”為檢索詞檢索中國知網數據庫,共有9篇相關的中文文獻及1篇外文文獻。據不完全統計(不排除重復病例可能),結合本例患兒,國內外共報道38例RHOBTB2基因變異患兒,根據目前現有資料,各病例臨床首發癥狀為癲癇發作表現的有29例 (29/38,76.3%),癲癇發作類型各不相同,包括局灶性、復雜性和全身強直陣攣性癲癇發作。部分患者出現遲發性熱性驚厥、運動障礙(如共濟失調、肌張力障礙、陣發性舞蹈病樣運動和交替性兒童偏癱)。目前已發現RHOBTB2基因變異位點13個,其中12個為錯義變異、1個為無義變異。其中10個變異位點位于BTB結構域內(p.184~p.522)[12],分別為c.1448G>A(p.Arg483His)、c.1532G>A(p.Arg511Gln)、c.1531C>T(p.Arg511Trp)、c.1519C>T(p.Arg507Cys)、c.1528A>G(p.Asn510Asp)、c.1421C>G(p.Ala474Gly)、c.1531C>G(p.Arg511Gly)、c.280C>T(p.Arg94Cys)、c.717G>C(p.Trp239Cys)和c.722C>A(p.Ser241Tyr),3個變異位點位于非BTB結構域,分別為c.460C>T(p.Arg154*),c.103G>A(p.Glu35Lys)和c.1975A>G(p.Thr659Ala)。本文所報道患兒基因突變位點為c.1531C>T(p.Arg511Trp),目前國內外報道與本患兒相同位點的病例共計7例(包括本例),其臨床特征如表1所示[7,8,11,13]。結合現有文獻資料可推測RHOBTB2 c.1531C>T(p.Arg511Trp)位點變異患者的癲癇發作通常發生在嬰兒早期,癲癇發作類型以癲癇持續狀態多見[15]。
綜上所述,目前研究表明RHOBTB2變異會導致DEE的發生。該病多在多在嬰兒早期發病,主要表現為癲癇發作及發育遲緩,少數患兒也會表現為急性腦病。目前該病相關致病機制尚未明確,且由于病例數少,尚無法得出最有效的治療方法[15]。若急性腦病或癲癇發作患兒同時出現發育遲緩應考慮為DEE,盡早完善基因檢測以明確診斷。早期識別、早期干預可以改善發育遲緩情況,甚至延緩其進程,以達到改善患兒生活質量的目的。
利益沖突聲明 所有作者無利益沖突。
RHOBTB2基因與發育性癲癇性腦病-64型(developmental and epileptic encephalopathy-64,DEE-64)相關,DEE-64以癲癇發作、嚴重智力障礙、運動功能受損、出生后小頭畸形為主要表現。發育性和癲癇性腦病(developmental and epileptic encephalopathy,DEE)是一組臨床和遺傳異質的年齡依賴性神經系統疾病[1],其特征是在嬰兒期或兒童早期出現難治性癲癇發作,伴有精神運動發育遲緩或倒退。截至2023年3月,在線人類孟德爾遺傳(online mendelian inheritance in man,OMIM)數據庫收錄DEE致病基因已達110個,其中常染色體基因占比超過90%[2]。2016年首次報道RHOBTB2基因變異引起的一例以重度神經發育障礙為特征的DEE,2018年在RHOBTB2的broad-complex,tramtrack,and bric-à-brac(BTB)結構域區域中聚集的新生雜合錯義突變所致DEE[3]。本文報道一例RHOBTB2變異所致DEE病例,該患兒嬰兒期有多次抽搐病史,表現為全面性或局灶性癲癇發作及嬰兒期喂養困難,服用抗癲癇發作藥物治療效果良好,發作間期腦電圖(electroencephalogram,EEG)顯示多灶性放電,頭顱磁共振成像(magnetic resonance imaging,MRI)提示雙側額顳部腦外間隙增寬。該研究獲得濟寧醫學院附屬醫院醫學倫理委員會審核批準及患兒監護人知情同意。
病例資料 患兒 女,3月齡余。于2023年7月5日因“間斷抽搐8天”入院。患兒入院前8天出現抽搐,表現為意識喪失、呼之不應、雙目凝視、面色發紺、四肢抽搐、口吐泡沫,歷時1~6 min,1次/d~1次/h不等,抽搐間期精神可,抽搐緩解后完善腦電圖未恢復至基線狀態,無其他伴隨癥狀,病后予以適當補液治療。入院前再次出現頻繁抽搐,每次持續約1 h緩解,緩解后精神略差,進食不佳。初步診斷為“癲癇”。查體未見明顯發育異常,完善格塞爾發展量表(GESELL)結果提示發育商87(發育正常兒童發育商>70),未見明顯發育遲緩。初予以左乙拉西坦口服液及苯巴比妥片治療3周仍有抽搐,加用托吡酯后癲癇控制良好,后于2023年10月減停左乙拉西坦口服液,未在有明顯發作。5月齡余(2023年9月17日)隨訪完善GESELL表結果提示發育商43.6(<70,達發育遲緩診斷標準),存在發育遲緩,各部分能區均不同程度落后,豎頭欠佳,僅可咿呀發聲,無明顯小頭畸形表現,不會叫爸爸媽媽,復查腦電圖大致正常。1歲3月齡(2024年6月29日)末次隨訪,頭圍42.0 cm,體重8.0 kg,存在小頭畸形、全面發育落后表現,復查腦電圖大致正常。出生后因“黃疸”住院治療,期間輸注血液制品(具體不詳)。家族史未見異常。其他無特殊。
輔助檢查 血生化:電解質、肝功能、腎功能、血氨、乳酸正常; 血液及尿液代謝病篩查未見異常;酮體9.5 ng/dL。2023年7月6日頭顱MRI平掃+彌散加權成像+癲癇特殊序列示:雙側海馬對稱,未見明顯異常信號影。雙側大腦半球結構對稱,腦灰白質對比正常,腦實質內未見異常信號灶,幕下小腦半球及腦干信號未見明顯異常。雙側額顳部腦外間隙增寬,余各腦室、腦池、腦裂及腦溝未見異常,中線結構居中。結論:① 腦內結構未見明確異常信號;② 雙側額顳部腦外間隙增寬(圖1)。2023年6月29日腦電圖示:嬰兒正常范圍睡眠腦電圖、腦地形圖。2023年7月6日床旁視頻腦電圖示:異常嬰兒視頻腦電圖睡眠期雙側額區、中央區不典型尖波散發記錄到6次起源于雙側前頭部局灶性發作并泛化(圖2)。后多次復查均提示異常嬰兒床旁視頻腦電圖,多灶性放電(較前好轉)。2023年9月3日 6 h視頻腦電圖示:大致正常嬰兒視頻腦電圖。全外顯子基因測序示:患兒RHOBTB2基因:c.1531C>T即該基因
圖1
患兒頭顱磁共振成像平掃+彌散加權成像+癲癇特殊序列
圖2
患兒腦電圖
a. 發作初期;b. 發作期;c. 發作終止
圖3
全外顯子基因測序
討論 本例患兒以早發性癲癇發作、癲癇持續狀態、發育遲緩為主要表現,輔助檢查提示無明顯感染指征,生化、尿液代謝指標不支持代謝性疾病,顱腦MRI不支持結構性病變所致,結合基因檢測結果符合RHOBTB2基因相關性DEE。該基因是含 Rho 相關BTB 結構域的蛋白構成非典型RhoGTP酶的一個亞家族,在哺乳動物中以RHOBTB1、RHOBTB2和RHOBTB3為代表[5]。其中RHOBTB2在神經系統中表達最豐富。RHOBTB2基因隸屬于致病基因中的酶/酶調節劑中的信號轉導酶類別,位于人類染色體8p21.3,該基因編碼的蛋白質是一種小的非典型Rho GTP酶,含有一個GTPase 結構域和2個BTB結構域。它通過BTB結構域與Culli-3依賴性泛素連接酶復合物相互作用,介導其自身的泛素化,并招募其他底物到復合物中,是細胞信號傳遞相關基因,參與各種細胞活動,如轉錄調節和蛋白質降解等。研究表明,在RHOBTB2的BTB結構域編碼區聚集的新發錯義變異的個體表現為相當均勻、嚴重的DEE,包括早發性癲癇、智力障礙、小頭畸形、陣發性運動障礙和MRI異常,與本例患兒所出現的早發性癲癇、智力障礙、小頭畸形、陣發性運動障礙高度一致[6,7]。同時Straub等[8]研究發現,Cullin-3結合位點位于第二個BTB結構域但與該p.Arg511Trp變體的位點相距甚遠,該基因位點突變不會破壞RHOBTB2的Cullin-3結合位點,且在既往報道的28例表型包括癲癇、中度至重度智力障礙的RHOBTB2新發錯義變異患者中,所有突變位點都聚集在或接近第一或第二BTB結構域,這更表明可能不是由于泛素連接酶支架蛋白cullin-3的直接相互作用介導的,而是由BTB結構域穩定性受損或二聚體形成介導的[7,9]。此外,敲低RHOBTB2基因表達量會導致樹突發育大小、長度顯著減少,導致突觸興奮與抑制失衡,RHOBTB2的過表達也會導致癲癇發作的易感性增加、運動障礙及增加促凋亡蛋白的水平,因此,DEE的病理機制也可能是由于突變體RHOBTB2水平增高增加了細胞凋亡所導致[6]。據統計,目前DEE的治療主要以對癥治療為主,多數患者口服2種或2種以上藥物治療癲癇和運動障礙,其中卡馬西平、左乙拉西坦等對運動障礙可能更有效[10],托吡酯和氟桂利嗪聯合用藥可能會降低運動障礙的發生頻率[11]。本例患兒的治療主要是口服苯巴比妥及托吡酯控制癲癇發作,目前病情控制良好,與既往文獻報道一致。
文獻復習顯示,截止2023年12月,以“(RHOBTB2)AND(Developmental epileptic encephalopathy)”為檢索詞條檢索Pubmed數據庫,共有6篇相關的英文文獻[7,8,11-14],以“RHOBTB2與癲癇”為檢索詞檢索中國知網數據庫,共有9篇相關的中文文獻及1篇外文文獻。據不完全統計(不排除重復病例可能),結合本例患兒,國內外共報道38例RHOBTB2基因變異患兒,根據目前現有資料,各病例臨床首發癥狀為癲癇發作表現的有29例 (29/38,76.3%),癲癇發作類型各不相同,包括局灶性、復雜性和全身強直陣攣性癲癇發作。部分患者出現遲發性熱性驚厥、運動障礙(如共濟失調、肌張力障礙、陣發性舞蹈病樣運動和交替性兒童偏癱)。目前已發現RHOBTB2基因變異位點13個,其中12個為錯義變異、1個為無義變異。其中10個變異位點位于BTB結構域內(p.184~p.522)[12],分別為c.1448G>A(p.Arg483His)、c.1532G>A(p.Arg511Gln)、c.1531C>T(p.Arg511Trp)、c.1519C>T(p.Arg507Cys)、c.1528A>G(p.Asn510Asp)、c.1421C>G(p.Ala474Gly)、c.1531C>G(p.Arg511Gly)、c.280C>T(p.Arg94Cys)、c.717G>C(p.Trp239Cys)和c.722C>A(p.Ser241Tyr),3個變異位點位于非BTB結構域,分別為c.460C>T(p.Arg154*),c.103G>A(p.Glu35Lys)和c.1975A>G(p.Thr659Ala)。本文所報道患兒基因突變位點為c.1531C>T(p.Arg511Trp),目前國內外報道與本患兒相同位點的病例共計7例(包括本例),其臨床特征如表1所示[7,8,11,13]。結合現有文獻資料可推測RHOBTB2 c.1531C>T(p.Arg511Trp)位點變異患者的癲癇發作通常發生在嬰兒早期,癲癇發作類型以癲癇持續狀態多見[15]。
綜上所述,目前研究表明RHOBTB2變異會導致DEE的發生。該病多在多在嬰兒早期發病,主要表現為癲癇發作及發育遲緩,少數患兒也會表現為急性腦病。目前該病相關致病機制尚未明確,且由于病例數少,尚無法得出最有效的治療方法[15]。若急性腦病或癲癇發作患兒同時出現發育遲緩應考慮為DEE,盡早完善基因檢測以明確診斷。早期識別、早期干預可以改善發育遲緩情況,甚至延緩其進程,以達到改善患兒生活質量的目的。
利益沖突聲明 所有作者無利益沖突。