

纖維多囊性肝病(fibropolycystic liver diseases,FLDs)是一類罕見的先天性膽管板畸形相關的遺傳性疾病,包括膽管錯構瘤、先天性肝纖維化、多囊性肝病、Caroli病及膽總管囊腫。該疾病臨床關注度低,影像學表現多樣,容易誤診和漏診。筆者通過本案例,總結該類疾病的典型影像學征象及其潛在的病理學機制,旨在加深讀者對該疾病的認識,從而降低該疾病的漏診率和誤診率。
引用本文: 汪媛媛, 羅春燕, 陳衛霞, 秦韻. 病例分析:纖維多囊性肝病的典型影像學表現. 中國普外基礎與臨床雜志, 2024, 31(9): 1059-1062. doi: 10.7507/1007-9424.202402024 復制
版權信息: ?四川大學華西醫院華西期刊社《中國普外基礎與臨床雜志》版權所有,未經授權不得轉載、改編
纖維多囊性肝病(fibropolycystic liver diseases,FLDs)是一類罕見的遺傳性疾病,包括一系列由膽管板畸形引起的累及肝臟和膽管系統的病變(膽管錯構瘤、先天性肝纖維化、多囊性肝病、Caroli病和膽總管囊腫) [1]。 FLDs的臨床表現多樣,患者可能沒有任何癥狀,也可能表現為膽道梗阻、膽管炎、門靜脈高壓和消化道出血相關的癥狀和體征[2]。目前針對該疾病的臨床關注度較低,其誤診率和漏診率較高。筆者希望通過總結FLDs的典型影像學征象及潛在的病理學機制,加深讀者對該疾病的認知和記憶,旨在提高該疾病的確診率。
1 患者基本信息
患者,女性,32歲。主因“發現肝內膽管擴張1+ 年,診斷重度貧血1+個月” ,為進一步診治就診于四川大學華西醫院(下文簡稱“我院” )。1+ 年前患者于外院檢查發現“肝內膽管囊性擴張”,無腹痛、腹脹、腹瀉、惡心、嘔吐,無黃疸、便血、食欲減退。就診我院查血常規:血紅蛋白 56 g/L,血小板計數 37×109/L。肝炎標志物(乙肝表面抗原及抗體、e抗原及抗體、核心抗體和丙肝)陰性,肝腎功能指標未見異常。腫瘤標志物:血清糖類抗原(carbohydrate antigen 19-9,CA19-9) 51.1 U/mL,其余腫瘤標志物(包括甲胎蛋白、CA125、癌胚抗原和異常凝血酶原)未見升高。患者一般情況良好,否認肝炎、結核或其他傳染病史,無吸煙史、無飲酒史。完善上腹部增強MRI檢查,未予特殊處理,定期于我院門診隨訪。1+ 個月前,患者偶感腹脹,進食后加重。查血常規、肝功、腎功、肝炎標志物和腫瘤標志物大致同前,行上腹部增強CT檢查。患者既往史及家族史無特殊。
2 影像學征象
患者上腹部增強MRI檢查提示:① 背景:肝硬化,門靜脈高壓伴門靜脈海綿樣變,脾臟增大;② 肝內多發不規則長T1長T2信號/低密度囊性灶,最大者長徑約2.5 cm,病灶主要沿肝內膽管走行,與膽管相通,未見彌散受限,增強掃描部分囊壁強化伴“中央點征” (圖1);③ 肝內多發長T1長T2信號/低密度小結節,不與膽管相通,未見彌散受限和異常強化,結節長徑均小于1.5 cm(圖2);④ 雙腎形態不規則伴雙側腎盞輕-中度擴張,雙腎多發長T1長T2信號結節,未見強化(圖2)。
 圖1
示該患者上腹部增強 MR及CT 檢查圖像顯示的Caroli病灶
圖1
示該患者上腹部增強 MR及CT 檢查圖像顯示的Caroli病灶
a:上腹部MR 軸位 T1 加權成像見肝內長T1信號結節(白箭示);b和c:軸位(b)及冠狀位(c) T2 加權脂肪抑制成像見肝內長T2信號結節,內見條狀低信號(白箭示);d和e:MR 軸位高 b 值(b=1 000)彌散加權成像(d)及表觀彌散系數圖(e),肝內病灶未見彌散受限(白箭示);f~h:MR軸位及冠狀位T1加權增強圖像,動脈期(f)、門靜脈期(g)及延遲期(h)見“中央點征”(白箭示病灶);i:CT增強掃描門靜脈期見低密度灶,內見強化血管影穿行(白箭示),并可見肝硬化改變及脾臟增大
 圖2
示該患者上腹部增強 MR及CT 檢查圖像顯示的錯構瘤和其他病變
圖2
示該患者上腹部增強 MR及CT 檢查圖像顯示的錯構瘤和其他病變
a:上腹部MR 軸位 T1 加權成像見肝內多發長T1信號小結節;b和c:軸位(b)及冠狀位(c) T2 加權脂肪抑制成像見肝內多發長T2信號小結節,雙腎形態不規則伴多發長T2信號結節,雙腎盞可見擴張;d和e:MR 軸位高 b 值(b=1 000)彌散加權成像(d)及表觀彌散系數圖(e),肝內病灶未見彌散受限;f~h:MR軸位及冠狀位T1加權增強圖像,動脈期(f)、門靜脈期(g)及延遲期(h),肝內及雙腎病灶未見強化;i:CT增強掃描門脈期肝內多發無強化低密度小結節
本例患者為青年女性,無肝炎、代謝及腫瘤病史,影像學檢查示肝硬化伴Caroli病、膽管錯構瘤,門靜脈高壓,脾臟增大,雙腎形態失常伴雙腎多發囊性改變,符合纖維多囊性肝病表現。
3 病理學檢查結果
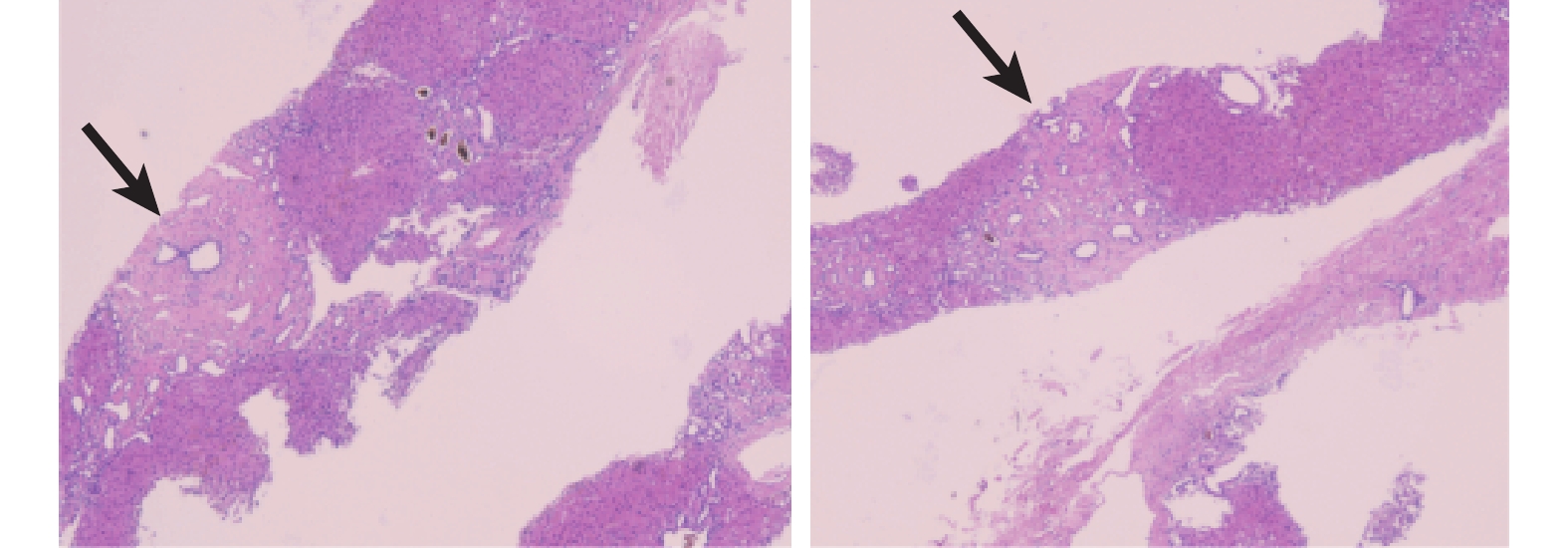
患者行肝右后葉穿刺,病理學檢查結果(圖3):共查見15個門管區;部分肝細胞內見脂褐素聚集;肝小葉界板較完整,壞死不明顯;匯管區見個別淋巴細胞及單核細胞浸潤;可見匯管區彌漫性纖維化形成纖維分隔,分割肝實質,纖維帶內及周邊見多數大小不一的膽管,部分膽管管腔擴張伴膽汁淤積。普魯士藍染色、羅丹寧染色及消化PAS染色均未見異常。Foot染色和Masson染色見寬大纖維隔形成。免疫組化結果:HBsAg(–)、HBcAg(–)、CD38(個別漿細胞+)、IgG4(–)。組織形態學表現為肝硬化伴膽管增生及粗大纖維形成。病理診斷符合先天性肝纖維化 (congenital hepatic fibrosis,CHF)。 需結合臨床、影像學及多囊腎和肝病基因1 (polycystic kidney and hepatic disease 1,PKHD1)檢測結果等綜合評價病因。
 圖3
示該患者肝臟穿刺活檢組織病理學檢查結果(HE ×40),見肝纖維化改變(黑箭示)
圖3
示該患者肝臟穿刺活檢組織病理學檢查結果(HE ×40),見肝纖維化改變(黑箭示)
4 討論
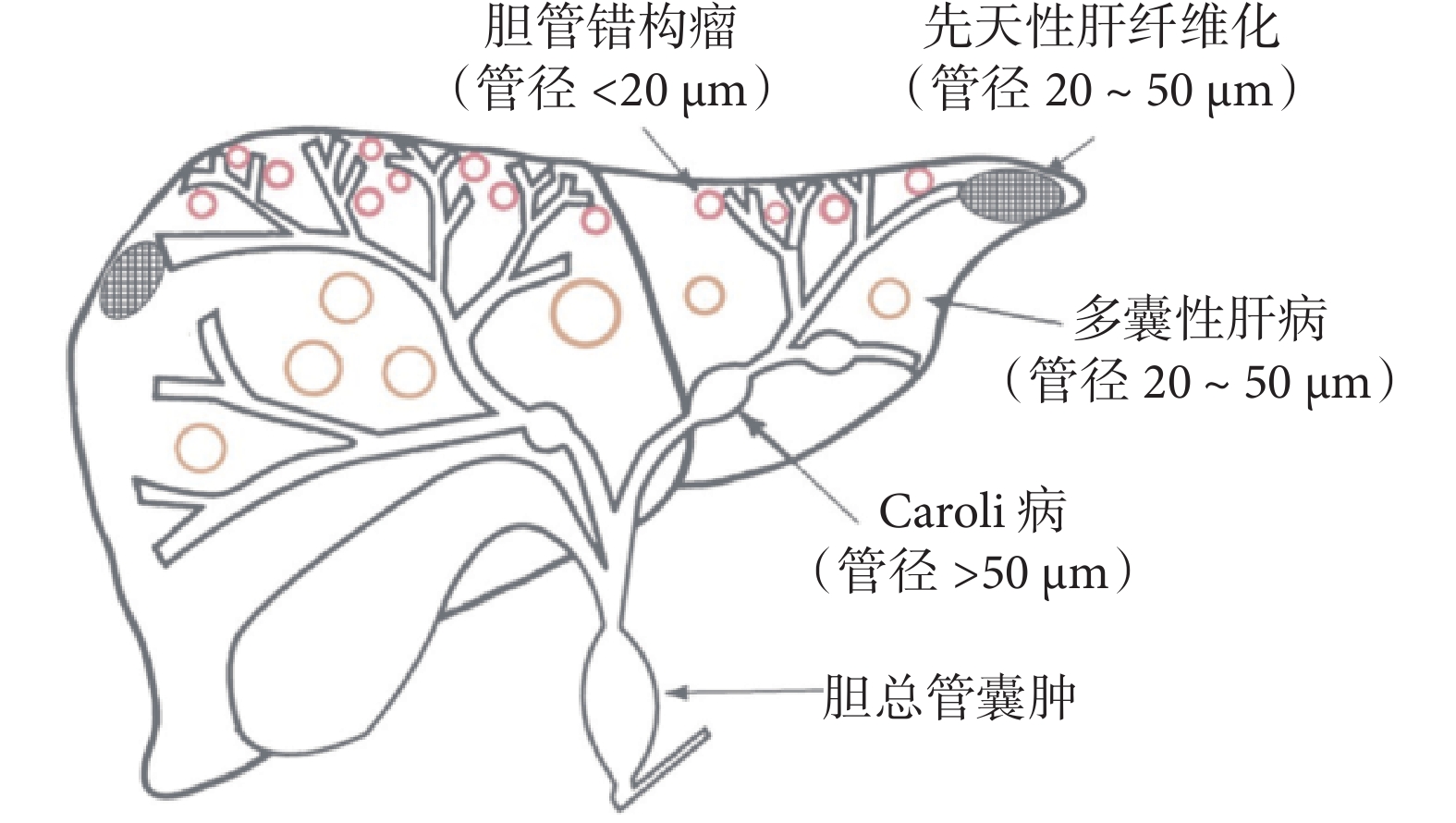
FLDs 是一類罕見的遺傳性疾病,部分患者是由PKHD1基因突變引起的,其病理基礎為膽管板發育畸形而導致的一系列疾病,包括膽管錯構瘤、先天性肝纖維化、多囊性肝病、Caroli病和膽總管囊腫(圖4)。以下將詳細介紹這些疾病的典型影像學征象。
 圖4
示膽管板畸形的類型
圖4
示膽管板畸形的類型
4.1 膽管錯構瘤
膽管錯構瘤,也稱為膽管微錯構瘤或 von Meyenburg 復合體(von Meyenburg complexes,VMCs),為內襯膽管上皮的一個或多個擴張管狀結構伴不同數量的纖維基質構成。該疾病主要起源于小葉內膽管(小型膽管,管徑 <20 μm)。膽管錯構瘤的典型影像學表現為肝內多發的圓形或不規則形狀的囊性病灶,大小相對均勻,直徑范圍為0.1~1.5 cm,與肝內膽管不相通。CT平掃病灶呈低密度,T1WI呈低信號,T2WI呈高信號。增強掃描病灶通常無強化,部分可見邊緣強化(病灶對鄰近肝實質的壓迫所致),無中央點狀強化[3-5]。
4.2 先天性肝纖維化
先天性肝纖維化是由PKHD1基因突變引起的,以青春期或成年早期患者多見。先天性肝纖維化組織病理學表現為門靜脈周圍/膽管周圍纖維化和膽管的不規則增生,以小葉間膽管和間隔膽管受累多見(中型膽管,管徑為20~50 μm)。先天性肝纖維化的典型影像學表現為肝臟的形態學改變,如左外葉和尾狀葉體積增大、左內葉正常或增大、右葉萎縮,而晚期病毒性和酒精性肝硬化則表現為左內葉體積減小。此外,該疾病常合并多囊腎病,多囊腎病的病理基礎為腎集合管非阻塞性梭狀擴張,其影像學表現為腎臟增大伴多發囊性灶[6-13]。
4.3 多囊性肝病
多囊性肝病是一種常染色體顯性遺傳疾病。該疾病涉及的突變基因包括PRKCSH、SEC63、GANAB、ALG8、PKD1、PKD2和PKHD1。該疾病是由小葉間膽管和間隔膽管的膽管板畸形(中型膽管,管徑為20~50 μm)導致的膽管囊狀擴張,隨著疾病進展,擴張的膽管逐漸與膽管束分離。多囊性肝病的典型影像學表現為肝臟均勻增大,伴多發大小不等的囊性灶,直徑多為0.1~12 cm,部分病灶直徑更大,與肝內膽管不相通,部分囊壁伴鈣化。CT平掃病灶呈低密度,T1WI呈低信號,T2WI呈高信號[3-4, 11, 14-15]。
4.4 Caroli病
Caroli病是由于肝內膽管的膽管板重塑受阻而導致的膽管囊狀或梭形擴張,累及大型膽管(管徑 >50 μm)。Caroli病的典型影像學表現為肝內膽管的多灶性、節段性擴張,以囊狀或梭形擴張為主,直徑可達5 cm,病變與膽管相通,常伴結石或膽淤泥 。MRCP有助于顯示囊狀或梭形擴張的膽管與膽道系統相通,用于與多囊性肝病和膽管錯構瘤等疾病鑒別。CT或MR增強掃描擴張膽管內常見纖維血管束走行,表現為“中央點征” 。Caroli病合并先天性肝纖維化被稱為Caroli綜合征[1, 16-17]。
5 總結
本例患者為青年女性,無肝炎、代謝及惡性腫瘤病史,CA19-9輕度升高,影像學檢查提示肝硬化及其繼發改變,同時合并Caroli病及膽管錯構瘤,且伴有雙側多囊腎表現,肝穿刺組織檢查結果提示先天性肝纖維化,符合FLDs改變。建議患者進一步完善PKHD1基因檢測,有助于后續精準治療方案制定。
重要聲明
利益沖突聲明: 本文全體作者閱讀并理解了《中國普外基礎與臨床雜志》的政策聲明,我們沒有相互競爭的利益。
作者貢獻聲明:汪媛媛和羅春燕查閱文獻、撰寫及修改論文;陳衛霞和秦韻審閱及修改論文。
倫理聲明:本研究通過了四川大學華西醫院生物醫學倫理審查委員會的審批,批文編號:2022年審(1487)號。
纖維多囊性肝病(fibropolycystic liver diseases,FLDs)是一類罕見的遺傳性疾病,包括一系列由膽管板畸形引起的累及肝臟和膽管系統的病變(膽管錯構瘤、先天性肝纖維化、多囊性肝病、Caroli病和膽總管囊腫) [1]。 FLDs的臨床表現多樣,患者可能沒有任何癥狀,也可能表現為膽道梗阻、膽管炎、門靜脈高壓和消化道出血相關的癥狀和體征[2]。目前針對該疾病的臨床關注度較低,其誤診率和漏診率較高。筆者希望通過總結FLDs的典型影像學征象及潛在的病理學機制,加深讀者對該疾病的認知和記憶,旨在提高該疾病的確診率。
1 患者基本信息
患者,女性,32歲。主因“發現肝內膽管擴張1+ 年,診斷重度貧血1+個月” ,為進一步診治就診于四川大學華西醫院(下文簡稱“我院” )。1+ 年前患者于外院檢查發現“肝內膽管囊性擴張”,無腹痛、腹脹、腹瀉、惡心、嘔吐,無黃疸、便血、食欲減退。就診我院查血常規:血紅蛋白 56 g/L,血小板計數 37×109/L。肝炎標志物(乙肝表面抗原及抗體、e抗原及抗體、核心抗體和丙肝)陰性,肝腎功能指標未見異常。腫瘤標志物:血清糖類抗原(carbohydrate antigen 19-9,CA19-9) 51.1 U/mL,其余腫瘤標志物(包括甲胎蛋白、CA125、癌胚抗原和異常凝血酶原)未見升高。患者一般情況良好,否認肝炎、結核或其他傳染病史,無吸煙史、無飲酒史。完善上腹部增強MRI檢查,未予特殊處理,定期于我院門診隨訪。1+ 個月前,患者偶感腹脹,進食后加重。查血常規、肝功、腎功、肝炎標志物和腫瘤標志物大致同前,行上腹部增強CT檢查。患者既往史及家族史無特殊。
2 影像學征象
患者上腹部增強MRI檢查提示:① 背景:肝硬化,門靜脈高壓伴門靜脈海綿樣變,脾臟增大;② 肝內多發不規則長T1長T2信號/低密度囊性灶,最大者長徑約2.5 cm,病灶主要沿肝內膽管走行,與膽管相通,未見彌散受限,增強掃描部分囊壁強化伴“中央點征” (圖1);③ 肝內多發長T1長T2信號/低密度小結節,不與膽管相通,未見彌散受限和異常強化,結節長徑均小于1.5 cm(圖2);④ 雙腎形態不規則伴雙側腎盞輕-中度擴張,雙腎多發長T1長T2信號結節,未見強化(圖2)。
圖1
示該患者上腹部增強 MR及CT 檢查圖像顯示的Caroli病灶
a:上腹部MR 軸位 T1 加權成像見肝內長T1信號結節(白箭示);b和c:軸位(b)及冠狀位(c) T2 加權脂肪抑制成像見肝內長T2信號結節,內見條狀低信號(白箭示);d和e:MR 軸位高 b 值(b=1 000)彌散加權成像(d)及表觀彌散系數圖(e),肝內病灶未見彌散受限(白箭示);f~h:MR軸位及冠狀位T1加權增強圖像,動脈期(f)、門靜脈期(g)及延遲期(h)見“中央點征”(白箭示病灶);i:CT增強掃描門靜脈期見低密度灶,內見強化血管影穿行(白箭示),并可見肝硬化改變及脾臟增大
圖2
示該患者上腹部增強 MR及CT 檢查圖像顯示的錯構瘤和其他病變
a:上腹部MR 軸位 T1 加權成像見肝內多發長T1信號小結節;b和c:軸位(b)及冠狀位(c) T2 加權脂肪抑制成像見肝內多發長T2信號小結節,雙腎形態不規則伴多發長T2信號結節,雙腎盞可見擴張;d和e:MR 軸位高 b 值(b=1 000)彌散加權成像(d)及表觀彌散系數圖(e),肝內病灶未見彌散受限;f~h:MR軸位及冠狀位T1加權增強圖像,動脈期(f)、門靜脈期(g)及延遲期(h),肝內及雙腎病灶未見強化;i:CT增強掃描門脈期肝內多發無強化低密度小結節
本例患者為青年女性,無肝炎、代謝及腫瘤病史,影像學檢查示肝硬化伴Caroli病、膽管錯構瘤,門靜脈高壓,脾臟增大,雙腎形態失常伴雙腎多發囊性改變,符合纖維多囊性肝病表現。
3 病理學檢查結果
患者行肝右后葉穿刺,病理學檢查結果(圖3):共查見15個門管區;部分肝細胞內見脂褐素聚集;肝小葉界板較完整,壞死不明顯;匯管區見個別淋巴細胞及單核細胞浸潤;可見匯管區彌漫性纖維化形成纖維分隔,分割肝實質,纖維帶內及周邊見多數大小不一的膽管,部分膽管管腔擴張伴膽汁淤積。普魯士藍染色、羅丹寧染色及消化PAS染色均未見異常。Foot染色和Masson染色見寬大纖維隔形成。免疫組化結果:HBsAg(–)、HBcAg(–)、CD38(個別漿細胞+)、IgG4(–)。組織形態學表現為肝硬化伴膽管增生及粗大纖維形成。病理診斷符合先天性肝纖維化 (congenital hepatic fibrosis,CHF)。 需結合臨床、影像學及多囊腎和肝病基因1 (polycystic kidney and hepatic disease 1,PKHD1)檢測結果等綜合評價病因。
圖3
示該患者肝臟穿刺活檢組織病理學檢查結果(HE ×40),見肝纖維化改變(黑箭示)
4 討論
FLDs 是一類罕見的遺傳性疾病,部分患者是由PKHD1基因突變引起的,其病理基礎為膽管板發育畸形而導致的一系列疾病,包括膽管錯構瘤、先天性肝纖維化、多囊性肝病、Caroli病和膽總管囊腫(圖4)。以下將詳細介紹這些疾病的典型影像學征象。
圖4
示膽管板畸形的類型
4.1 膽管錯構瘤
膽管錯構瘤,也稱為膽管微錯構瘤或 von Meyenburg 復合體(von Meyenburg complexes,VMCs),為內襯膽管上皮的一個或多個擴張管狀結構伴不同數量的纖維基質構成。該疾病主要起源于小葉內膽管(小型膽管,管徑 <20 μm)。膽管錯構瘤的典型影像學表現為肝內多發的圓形或不規則形狀的囊性病灶,大小相對均勻,直徑范圍為0.1~1.5 cm,與肝內膽管不相通。CT平掃病灶呈低密度,T1WI呈低信號,T2WI呈高信號。增強掃描病灶通常無強化,部分可見邊緣強化(病灶對鄰近肝實質的壓迫所致),無中央點狀強化[3-5]。
4.2 先天性肝纖維化
先天性肝纖維化是由PKHD1基因突變引起的,以青春期或成年早期患者多見。先天性肝纖維化組織病理學表現為門靜脈周圍/膽管周圍纖維化和膽管的不規則增生,以小葉間膽管和間隔膽管受累多見(中型膽管,管徑為20~50 μm)。先天性肝纖維化的典型影像學表現為肝臟的形態學改變,如左外葉和尾狀葉體積增大、左內葉正常或增大、右葉萎縮,而晚期病毒性和酒精性肝硬化則表現為左內葉體積減小。此外,該疾病常合并多囊腎病,多囊腎病的病理基礎為腎集合管非阻塞性梭狀擴張,其影像學表現為腎臟增大伴多發囊性灶[6-13]。
4.3 多囊性肝病
多囊性肝病是一種常染色體顯性遺傳疾病。該疾病涉及的突變基因包括PRKCSH、SEC63、GANAB、ALG8、PKD1、PKD2和PKHD1。該疾病是由小葉間膽管和間隔膽管的膽管板畸形(中型膽管,管徑為20~50 μm)導致的膽管囊狀擴張,隨著疾病進展,擴張的膽管逐漸與膽管束分離。多囊性肝病的典型影像學表現為肝臟均勻增大,伴多發大小不等的囊性灶,直徑多為0.1~12 cm,部分病灶直徑更大,與肝內膽管不相通,部分囊壁伴鈣化。CT平掃病灶呈低密度,T1WI呈低信號,T2WI呈高信號[3-4, 11, 14-15]。
4.4 Caroli病
Caroli病是由于肝內膽管的膽管板重塑受阻而導致的膽管囊狀或梭形擴張,累及大型膽管(管徑 >50 μm)。Caroli病的典型影像學表現為肝內膽管的多灶性、節段性擴張,以囊狀或梭形擴張為主,直徑可達5 cm,病變與膽管相通,常伴結石或膽淤泥 。MRCP有助于顯示囊狀或梭形擴張的膽管與膽道系統相通,用于與多囊性肝病和膽管錯構瘤等疾病鑒別。CT或MR增強掃描擴張膽管內常見纖維血管束走行,表現為“中央點征” 。Caroli病合并先天性肝纖維化被稱為Caroli綜合征[1, 16-17]。
5 總結
本例患者為青年女性,無肝炎、代謝及惡性腫瘤病史,CA19-9輕度升高,影像學檢查提示肝硬化及其繼發改變,同時合并Caroli病及膽管錯構瘤,且伴有雙側多囊腎表現,肝穿刺組織檢查結果提示先天性肝纖維化,符合FLDs改變。建議患者進一步完善PKHD1基因檢測,有助于后續精準治療方案制定。
重要聲明
利益沖突聲明: 本文全體作者閱讀并理解了《中國普外基礎與臨床雜志》的政策聲明,我們沒有相互競爭的利益。
作者貢獻聲明:汪媛媛和羅春燕查閱文獻、撰寫及修改論文;陳衛霞和秦韻審閱及修改論文。
倫理聲明:本研究通過了四川大學華西醫院生物醫學倫理審查委員會的審批,批文編號:2022年審(1487)號。